bachelorstudiengang molekulare biotechnologie … · in the present bachelor thesis a cellular...
TRANSCRIPT

Etablierung eines zellulären in vitro Assays zur Bestimmung der
Angiotensin II Aktivität basierend auf der Expression von Luziferase
durch die Aktivierung von NF-κB
2. Bachelorarbeit
Bachelorstudiengang Molekulare Biotechnologie
Fachhochschule Campus Wien
Vorgelegt von:
Rebecca Petri
Personenkennzeichen: 0710543035
Betreuer:
Dr. Günther Staffler
Abgabetermin: 04.06.2010

Kurzfassung
Etablierung eines zellulären in vitro Assays zur Bestimmung der Angiotensin II
Aktivität basierend auf der Expression von Luziferase durch die Aktivierung von
NF-κB
Arterielle Hypertonie, allgemein bekannt als Bluthochdruck, erhöht das Risiko für
kardiovaskuläre Krankheiten drastisch und stellt daher in der heutigen Gesellschaft ein
zunehmendes Problem dar.
Im Körper spielt das Renin-Angiotensin System (RAS) eine entscheidende Rolle in der
Regulierung des Blutdruckes. Die physiologisch aktivste Komponente dieses Systems ist
das Peptid-Hormon Angiotensin II (AngII). AngII bindet an den Angiotensin II Typ 1
Rezeptor (AT1R) und bewirkt dadurch die Verengung von Arteriolen, die Freisetzung von
Aldosteron und die renale Aufnahme von Natrium, alles Faktoren, die zu einer
Blutdrucksteigerung führen. Unter pathophysiologischen Bedingungen ist AngII
maßgeblich an der Entwicklung eines chronisch erhöhten Blutdruckes beteiligt. Somit
stellt das Renin-Angiotensin System ein sehr gutes Angriffsziel für Therapien gegen
arterielle Hypertonie dar.
Die vorliegende Bachelorarbeit ist Teil eines Projektes von AFFiRiS AG, dessen Ziel es
ist, einen Peptidimpfstoff zu entwickeln, welcher Angiotensin II (AngII) neutralisiert und da
durch für die Behandlung von Bluthochdruck eingesetzt werden kann. Im Zuge dieser
Bachelorarbeit soll ein zellulärer in vitro Assay entwickelt werden, mit dem die Messung
der AngII Aktivität möglich ist. Dieser zelluläre Assay basiert auf der transienten
Kotransfektion eines Angiotensin II Typ 1 Rezeptor-„Enhanced Green Fluorescent
Protein“ (AT1R-EGFP) Expressionsvektors und eines Nukleärer Faktor-κB Luziferase
Reporter Gen System (NF-κB-luc) Expressionsvektors in menschliche Nierenzellen
(HEK293). Die Zellen werden nach der Transfektion mit unterschiedlichen AngII
Konzentrationen stimuliert. Durch die Bindung von AngII an den AT1 Rezeptor wird NF-κB
freigesetzt. Dieser bindet an das Luziferase Response Element und löst so die Produktion
von Luziferase aus. Es konnte gezeigt werden, dass AngII zu einer erhöhten
Luziferaseproduktion führt und diese durch monoklonale Antikörper gehemmt werden
kann.
2

Abstract
Establishment of a cellular in vitro assay for the evaluation of angiotensinII activity
based on the expression of luciferase triggered by the activation of NF-κB
High blood pressure increases the risk of cardiovascular diseases dramatically and
therefore represents a severe problem in today´s society. The renin-angiotensin system
(RAS) plays a major role in the control of blood pressure. The physiologically most active
component of this system is the peptide hormone angiotensin II (AngII). Its binding to the
angiotensin II type 1 receptor (AT1R) promotes vascular resistance, the release of
aldosteron and the renal reabsorption of sodium. Under pathological conditions all those
functions lead to a chronically elevated blood pressure. Due to its function and due to the
fact that different drugs exist that interfere with the RAS, this system represents a well
characterized and validated target for the treatment of hypertension.
The present bachelor thesis is part of a project of AFFiRiS AG. AFFiRiS AG aims at
developing a vaccine that neutralizes AngII and thus lowers blood pressure in
hypertensive patients. In the present bachelor thesis a cellular assay is developed which
allows the measurement of AngII activity. The cellular assay is based on the co
transfection of an angiotensin II type 1 receptor-Enhanced Green Fluorescent Protein
(AT1R- EGFP) vector and a nuclear factor-κB luciferase reporter gene system (NF-κB-luc)
vector into Human Embryonic Kidney (HEK293) cells. Upon transfection HEK293 cells are
stimulated by different concentrations of AngII. The binding of AngII to the AT1 receptor
triggers the release of NF-κB. Subsequently NF-κB binds to the luciferase response
element and leads to the expression of luciferase. In the present work we were able to
show that AngII leads to an increased luciferase production which can be inhibited by an
AngII-specific monoclonal antibody.
3

Inhaltsverzeichnis
1. Einleitung....................................................................................................................5
2. Ergebnisse .................................................................................................................9
2.1 Transfektion von HEK293 Zellen mit einem EGFP-N1 Vektor ..................................9
2.2 Testung unterschiedlicher Transfektionsmethoden ................................................11
2.3 Überprüfung der Plasmid DNA mittels Restriktionsfragmentanalyse ......................12
2.4 Transfektionen unterschiedlicher DNA-Konstrukte .................................................13
2.5 Bestimmung der Kinetik der transienten Transfektion.............................................14
2.6 Lokalisierung des AT1 Rezeptors mittels Fluoreszenzmikroskopie .........................15
2.7 Stimulation der HEK293 Zellen und Messung der Luziferaseproduktion ................16
2.8 Testung monoklonaler Antikörper...........................................................................18
3. Diskussion ................................................................................................................19
4. Literaturverzeichnis ..................................................................................................22
5. Eigenständigkeitserklärung.......................................................................................23
6. Anhang.....................................................................................................................24
6.1 Materialien und Methoden......................................................................................24
6.2 Abkürzungsverzeichnis ..........................................................................................29
6.3 Abbildungsverzeichnis............................................................................................30
6.4 Protokolle ...............................................................................................................31
4

1. Einleitung
Arterielle Hypertonie, allgemein bekannt als Bluthochdruck, stellt in der heutigen
Gesellschaft ein schwerwiegendes Problem dar, da sie das Risiko für Herz-
Kreislauferkrankungen und deren Begleiterscheinungen, wie Schlaganfälle, Nieren
fehlfunktionen und Herzrhythmusstörungen drastisch erhöht.1 Die Entstehung von
Bluthochdruck kann von vielen Faktoren beeinflusst und ausgelöst werden. Ernährung,
Fettleibigkeit, Bewegungsmangel und genetische Veranlagung stellen potentielle
Risikofaktoren für die Entstehung arterieller Hypertonie dar.2
Im Körper spielt das Renin-Angiotensin System (RAS) eine bedeutende Rolle in der
Kontrolle des Blutdruckes. Renin ist eine Peptidase, die vorwiegend in den
juxtaglomerulären Zellen der Niere produziert wird. Sie spaltet das in der Leber produ
zierte Protein Angiotensinogen, wodurch das Dekapeptid Angiotensin I (AngI) entsteht.
Durch die Abspaltung zweier Aminosäuren von AngI durch das Angiotensin Converting
Enzyme (ACE) wird das Peptid-Hormon Angiotensin II (AngII) gebildet. Angiotensin II ist
acht Aminosäuren lang und ist die physiologisch aktivste Komponente des RA-Systems.
Es wird kurz nach seiner Produktion von Angiotensinasen weiter zu Angiotensin III
(AngIII) und Angiotensin IV (AngIV) abgebaut.1
AngII kann an die spezifischen Zelloberflächen-Rezeptoren, den Angiotensin II Typ 1
(AT1) Rezeptor und den Angiotensin II Typ 2 (AT2) Rezeptor binden und so
unterschiedliche biologische Funktionen erfüllen (vgl. Abb. 1). Beide Rezeptoren gehören
der Klasse der „G-Protein Coupled Receptors“ (GPCR) an und besitzen eine Sieben
Transmembran-Domäne.3
Der AT1 Rezeptor ist auf zahlreichen Geweben im Körper lokalisiert. Neben der glatten
Gefäßmuskulatur wird der AT1 Rezeptor auch in der Niere, in der Nebenniere, im Gehirn,
und im Herzen exprimiert.3 Die Bindung von AngII an den AT1 Rezeptor induziert unter
anderem die Verengung von Arteriolen, die verstärkte Freisetzung von Aldosteron und die
Aufnahme von Natrium in die Niere.1 All diese Faktoren führen zu einer Erhöhung des
Blutdruckes. Unter pathophysiologischen Bedingungen steht die Bindung von AngII an
den AT1R in direkter Verbindung mit der Entstehung von chronischer, arterieller
Hypertonie. Bei der Bindung von Ang II an den AT1 Rezeptor wird das daran gekoppelte
G-Protein aktiviert. Dieses aktiviert wiederum unter anderem die Phospholipase C (PLC),
welche Phosphatidylinositol-4,5-Bisphosphat (PIP2) in Inositol-1,4,5-triphosphat (IP3) und
Diacylglycerol (DAG) spaltet. IP3 löst die Freisetzung von Calcium aus dem
5

Endoplasmatischen Retikulum aus. Dieser Calciumeinstrom führt zusammen mit DAG zur
Aktivierung verschiedener Enzyme, die Einfluss auf die Zellfunktion nehmen.4 Über diesen
Signalweg wird auch die Iκ-Kinase β aktiviert. Sie phosphoryliert I-κB, einen Inhibitor von
NF-κB, und löst so die Freisetzung des Transkriptionsfaktors aus.
Im Gegensatz zu der Funktion des AT1 Rezeptors ist jene des AT2 Rezeptors noch relativ
unbekannt. Es wird jedoch vermutet, dass er vielen Funktionen des AT1 Rezeptors
entgegensteuert. Im Fetus wird der AT2 Rezeptor in allen Geweben exprimiert. Nach der
Geburt erfolgt die Expression des Rezeptors in einigen Geweben nur noch in sehr gerin
gen Mengen oder wird gänzlich gestoppt.3 Experimente zeigen, dass die Bindung von
AngII an den AT2 Rezeptor zur Gefäßerweiterung und zur Freisetzung von
Stickstoffmonoxid führt. Außerdem werden Apoptose und Zelldifferenzierung gefördert
und Zellwachstum gehemmt. Es wird angenommen, dass die Signalweiterleitung des AT2
Rezeptors G-Protein abhängig und unabhängig über die Aktivierung von Serin/Threonin
und Tyrosin Phosphatasen geschieht.5
Abb. 1: Renin-Angiotensin Hormonkaskade. Renin spaltet Angiotensinogen in Angiotensin I. Dieses wird vom
Angiotensin Converting Enzyme (ACE) in Angiotensin II gespaltet, welches acht Aminosäuren lang ist. AngII kann
an den AT1 Rezeptor und AT2 Rezeptor binden und so unterschiedliche Funktionen induzieren.1
Da arterielle Hypertonie zu schwerwiegenden Folgen führen kann, wurden bereits
zahlreiche Medikamente und Therapien entwickelt, die diesen entgegensteuern. Da das
Renin-Angiotensin System ein attraktives Angriffsziel für Therapien darstellt, wurden be
reits ACE Inhibitoren, AT1R Blocker und Renin Inhibitoren gegen Bluthochdruck
entwickelt.4
AFFiRiS AG hat das Ziel, einen Impfstoff auf Peptidbasis zu entwickeln, der die
blutdruckerhöhende Wirkung von AngII verhindert. Hierzu verwendet AFFiRiS AG
6

VARIOTOPE®. VARIOTOPE® sind Peptide, die sich in ihrer Aminosäuresequenz von
AngII unterscheiden, jedoch trotzdem eine humorale Immunantwort gegen dieses Hormon
auslösen können. Die vorliegende Bachelorarbeit wurde im Rahmen dieses Projektes
verfasst. Das Ziel dieser Arbeit war einen zellulären Assay zu entwickeln, mit dem es
möglich ist, die inhibierende Wirkung der VARIOTOP®-induzierten Antikörper auf
Angiotensin II zu testen.
Abb. 2: Prinzip des zellulären Assays. Kommt es zu einer Bindung von AngII an den AT1 Rezeptor, wird in den
HEK293 Zellen NF-κB freigesetzt. Der Transkriptionsfaktor bindet an das „Response Element“ auf dem Vektor und
es k ommt zur Produktion von Luziferase. Dieses Signal kann gemessen werden.
Das Prinzip dieses Assays ist in Abbildung 2 dargestellt. Es basiert auf der Kotransfektion
eines NF-κB Luziferase Reporter Gen System (NF-κB-luc) Expressionsvektors und eines
AT1R-EGFP Expressionsvektors (vgl. Abb. 3) in menschliche Nierenzellen (HEK293).
Nach der Expression des AT1 Rezeptors an der Zelloberfläche, kann AngII an den AT1
Rezeptor binden. Durch diese Bindung wird I-κB, ein Inhibitor von NF-κB, durch die Iκ
Kinaseβ phosphoryliert. I-κB wird anschließend ubiquitinyliert und durch das Proteasom
abgebaut.6 NF-κB wird nun freigesetzt und löst durch die Bindung an das eingebrachte
Luziferase Response Element die Produktion von Luziferase aus. In der Gegenwart von
VARIOTOP®-induzierten Antikörpern, die freies AngII neutralisieren, sollte die
Freisetzung von NF-κB nicht erfolgen und daher kein Luziferasesignal messbar sein.
An Hand der unterschiedlichen Lumineszenzsignale sollte daher die inhibierende Wirkung
der VARIOTOP®-induzierten Antiköper auf AngII bewertet werden.
7

Abb. 3: AT1R-EGFP Expressionsvektor. Der Vektor trägt an seinem C-Terminus ein EGFP-Gen. Die AT1R cDNA
wurde in die „Multiple Cloning Site“ (MCS) am N-Terminus des EGFP Gens kloniert. Dies erlaubte die Expression
eines EGFP-AT1R Fusionsproteins.
Im Zuge dieser Bachelorarbeit sollten die Expressionsvektoren AT1R-EGFP und NF-κB
luc transient in HEK293 Zellen kotransfektiert werden und die Transfektionseffizienz durch
„Fluoresence Activated Cell Sorting“ (FACS) Analysen bestimmt werden. Um die
Funktionalität des Assays zu überprüfen sollten HEK293 Zellen nach der Kotransfektion
mit unterschiedlichen AngII Konzentrationen stimuliert und die Luziferaseproduktion
gemessen werden. In weiterer Folge sollte mit monoklonalen Antikörpern getestet
werden, ob die Aktivität von AngII inhibiert werden kann. Ziel dieser Bachelorarbeit war,
die Basis für zukünftige Versuche zu schaffen, in denen, mit Hilfe des Luziferase Assays,
die inhibierende Wirkung von VARIOTOP®-induzierten Antiköpern aus Seren getestet
werden kann.
8
2. Ergebnisse
2.1 Transfektion von HEK293 Zellen mit einem EGFP-N1 Vektor
Im Zuge der vorliegenden Bachelorarbeit sollte ein zellulärer Assay mit menschlichen
Nierenzellen (HEK293) etabliert werden, mit dem es möglich ist die inhibierende Wirkung
von VARIOTOP®-induzierten Antikörpern auf AngII zu messen.
Im ersten Schritt wurden HEK293 Zellen mit einem EGFP-N1 Vektor transfektiert, um zu
ermitteln, wie effizient diese Zellen transfektiert werden können. Die Zellen wurden in den
Dichten 0,25x106 Zellen/ml, 0,5x106 Zellen/ml und 1x106 Zellen/ml ausgesät. Die
Transfektion wurde mit Calcium Phosphat durchgeführt und 24 Stunden nach der
Transfektion die Transfektionseffizienz durch eine FACS Analyse bestimmt.
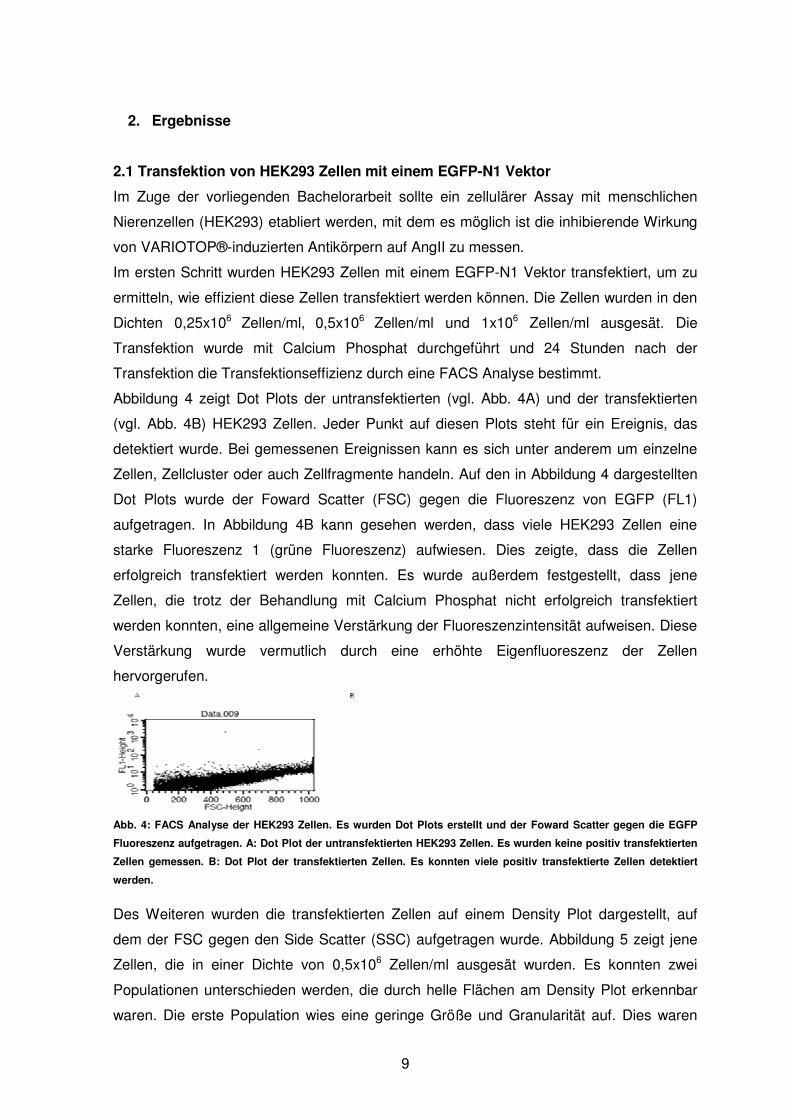
Abbildung 4 zeigt Dot Plots der untransfektierten (vgl. Abb. 4A) und der transfektierten
(vgl. Abb. 4B) HEK293 Zellen. Jeder Punkt auf diesen Plots steht für ein Ereignis, das
detektiert wurde. Bei gemessenen Ereignissen kann es sich unter anderem um einzelne
Zellen, Zellcluster oder auch Zellfragmente handeln. Auf den in Abbildung 4 dargestellten
Dot Plots wurde der Foward Scatter (FSC) gegen die Fluoreszenz von EGFP (FL1)
aufgetragen. In Abbildung 4B kann gesehen werden, dass viele HEK293 Zellen eine
starke Fluoreszenz 1 (grüne Fluoreszenz) aufwiesen. Dies zeigte, dass die Zellen
erfolgreich transfektiert werden konnten. Es wurde außerdem festgestellt, dass jene
Zellen, die trotz der Behandlung mit Calcium Phosphat nicht erfolgreich transfektiert
werden konnten, eine allgemeine Verstärkung der Fluoreszenzintensität aufweisen. Diese
Verstärkung wurde vermutlich durch eine erhöhte Eigenfluoreszenz der Zellen
hervorgerufen.
Abb. 4: FACS Analyse der HEK293 Zellen. Es wurden Dot Plots erstellt und der Foward Scatter gegen die EGFP
Fluoreszenz aufgetragen. A: Dot Plot der untransfektierten HEK293 Zellen. Es wurden keine positiv transfektierten
Zellen gemessen. B: Dot Plot der transfektierten Zellen. Es konnten viele positiv transfektierte Zellen detektiert
werden.
Des Weiteren wurden die transfektierten Zellen auf einem Density Plot dargestellt, auf
dem der FSC gegen den Side Scatter (SSC) aufgetragen wurde. Abbildung 5 zeigt jene
Zellen, die in einer Dichte von 0,5x106 Zellen/ml ausgesät wurden. Es konnten zwei
Populationen unterschieden werden, die durch helle Flächen am Density Plot erkennbar
waren. Die erste Population wies eine geringe Größe und Granularität auf. Dies waren
9
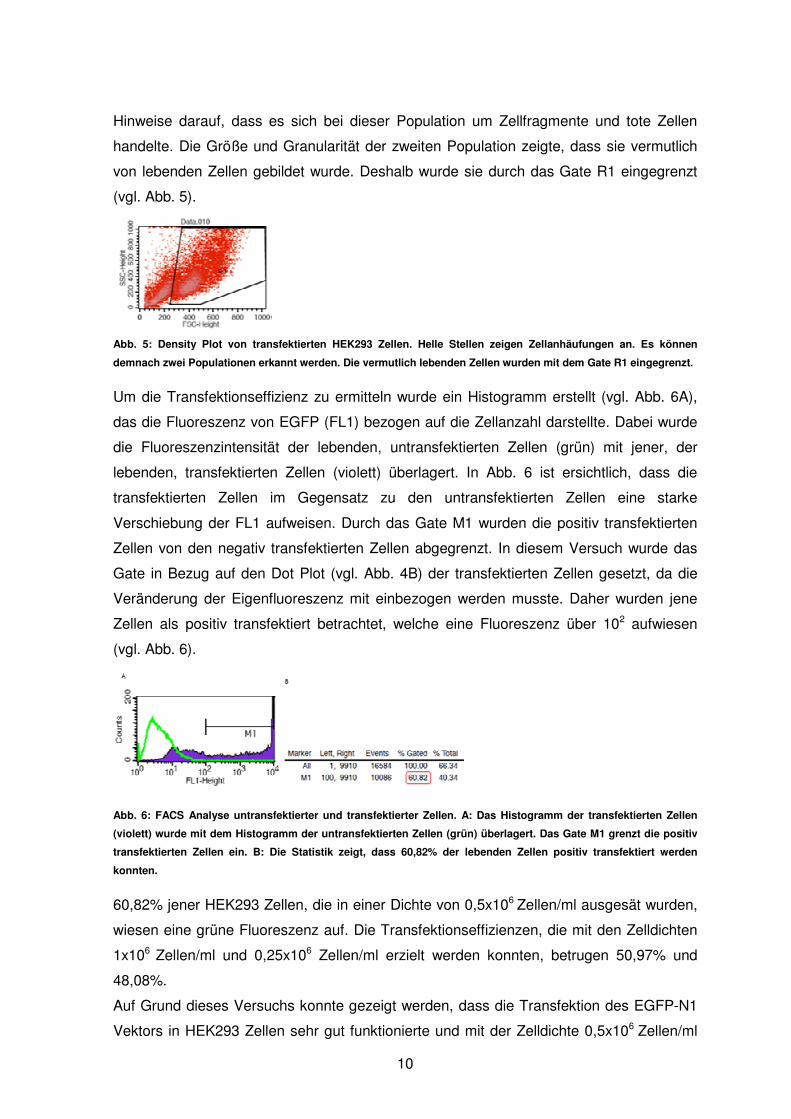
Hinweise darauf, dass es sich bei dieser Population um Zellfragmente und tote Zellen
handelte. Die Größe und Granularität der zweiten Population zeigte, dass sie vermutlich
von lebenden Zellen gebildet wurde. Deshalb wurde sie durch das Gate R1 eingegrenzt
(vgl. Abb. 5).
Abb. 5: Density Plot von transfektierten HEK293 Zellen. Helle Stellen zeigen Zellanhäufungen an. Es können
demnach zwei Populationen erkannt werden. Die vermutlich lebenden Zellen wurden mit dem Gate R1 eingegrenzt.
Um die Transfektionseffizienz zu ermitteln wurde ein Histogramm erstellt (vgl. Abb. 6A),
das die Fluoreszenz von EGFP (FL1) bezogen auf die Zellanzahl darstellte. Dabei wurde
die Fluoreszenzintensität der lebenden, untransfektierten Zellen (grün) mit jener, der
lebenden, transfektierten Zellen (violett) überlagert. In Abb. 6 ist ersichtlich, dass die
transfektierten Zellen im Gegensatz zu den untransfektierten Zellen eine starke
Verschiebung der FL1 aufweisen. Durch das Gate M1 wurden die positiv transfektierten
Zellen von den negativ transfektierten Zellen abgegrenzt. In diesem Versuch wurde das
Gate in Bezug auf den Dot Plot (vgl. Abb. 4B) der transfektierten Zellen gesetzt, da die
Veränderung der Eigenfluoreszenz mit einbezogen werden musste. Daher wurden jene
Zellen als positiv transfektiert betrachtet, welche eine Fluoreszenz über 102 aufwiesen
(vgl. Abb. 6).
Abb. 6: FACS Analyse untransfektierter und transfektierter Zellen. A: Das Histogramm der transfektierten Zellen
(violett) wurde mit dem Histogramm der untransfektierten Zellen (grün) überlagert. Das Gate M1 grenzt die positiv
transfektierten Zellen ein. B: Die Statistik zeigt, dass 60,82% der lebenden Zellen positiv transfektiert werden
konnten.
60,82% jener HEK293 Zellen, die in einer Dichte von 0,5x106 Zellen/ml ausgesät wurden,
wiesen eine grüne Fluoreszenz auf. Die Transfektionseffizienzen, die mit den Zelldichten
1x106 Zellen/ml und 0,25x106 Zellen/ml erzielt werden konnten, betrugen 50,97% und
48,08%.
Auf Grund dieses Versuchs konnte gezeigt werden, dass die Transfektion des EGFP-N1
Vektors in HEK293 Zellen sehr gut funktionierte und mit der Zelldichte 0,5x106 Zellen/ml
10

die höchste Transfektionseffizienz erreicht werden kann. Aus diesem Grund wurde diese
Zelldichte für weitere Versuche beibehalten.
2.2 Testung unterschiedlicher Transfektionsmethoden
Es gibt eine große Auswahl an unterschiedlichen Transfektionsreagenzien. Um jenes
Reagenz zu bestimmen, mit dem die höchste Transfektionseffizienz bei HEK293 Zellen
erreicht werden kann, wurden neben Calcium Phosphat, das FuGENE®6 Transfektions
reagenz von Roche, sowie das Lipofectamine™ Reagenz von Invitrogen getestet.
HEK293 Zellen wurden mit dem EGFP-N1 Vektor transfektiert und die jeweiligen
Transfektionseffizienzen durch FACS Analysen bestimmt.
Wie in Abbildung 7 ersichtlich, führten alle getesteten Transfektionsmethoden zu positiv
transfektierten Zellen (vgl. Abb. 7 A1, B1, C1). Die lebenden Zellen wurden in Density
Plots durch das Gate R1 eingegrenzt. Die Histogramme der transfektierten Zellen (violett)
wurden mit den Histogrammen der untransfektierten Zellen (grün) überlagert (vgl. Abb. 7
A2, B2, C2). Die positiv transfektierten Zellen wurden mit dem Gate M1 eingegrenzt (vgl.
Abb. 7 A2, B2, C2). Auch hier musste die allgemeine Veränderung der Eigenfluoreszenz
mit einbezogen werden, die vor allem bei den Transfektionen mit dem Lipofectamine™
Reagenz und Calcium Phosphat zu beobachten war. Es wurde ermittelt, dass mit
Lipofectamine™ die höchste Transfektionseffizienz erzielt werden konnte (~62%).
Allerdings wurde bei dieser Transfektion auch die höchste Zahl an toten Zellen gemessen.
Die Transfektion mit Calcium Phosphat war mit einer Transfektionseffizienz von 46,88%
ebenfalls sehr erfolgreich. Mit dem FuGENE®6 Transfektionsreagenz konnte allerdings
nur eine Transfektionseffizienz von 19,92% erzielt werden. Diese Methode schien daher
für die Transfektion und für HEK293 Zellen nicht optimal geeignet zu sein.
Auf Grund der hohen Transfektionseffizienz, der geringeren Rate an toten Zellen und
nicht zuletzt auf Grund der einfachen Handhabung wurde die Calcium Phosphat
Transfektionsmethode auch für alle weiteren Transfektionen angewandt.
11
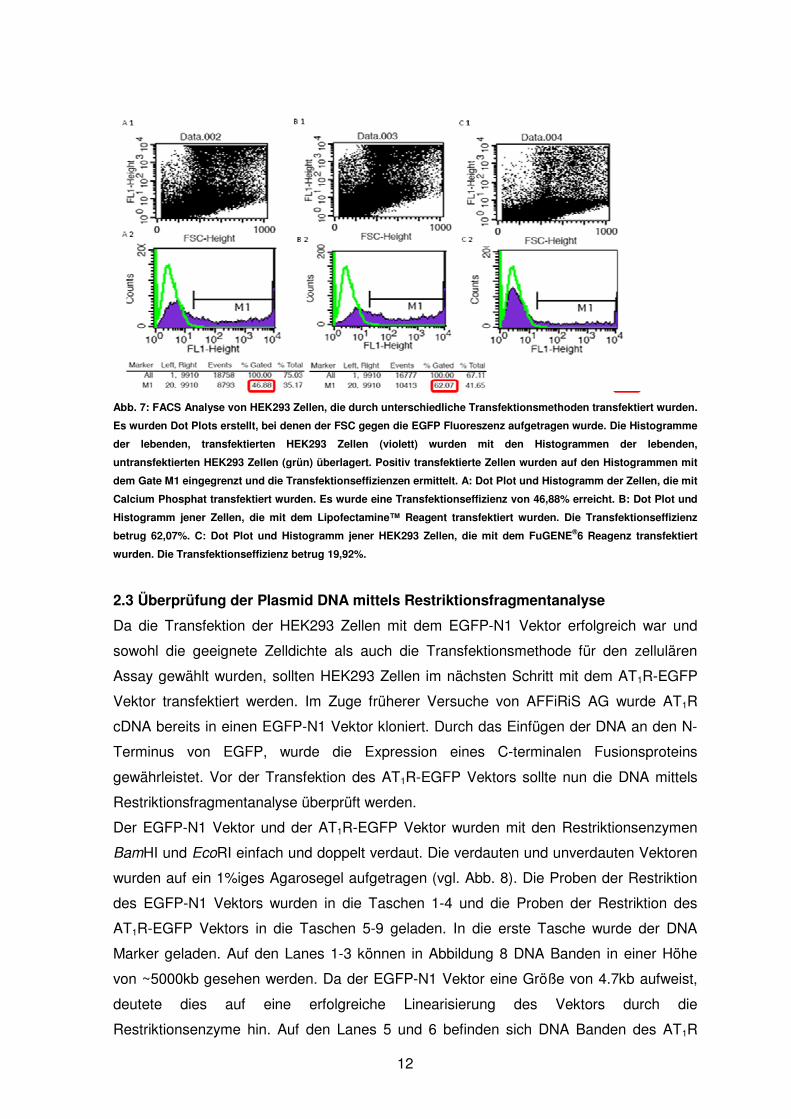
Abb. 7: FACS Analyse von HEK293 Zellen, die durch unterschiedliche Transfektionsmethoden transfektiert wurden.
Es wurden Dot Plots erstellt, bei denen der FSC gegen die EGFP Fluoreszenz aufgetragen wurde. Die Histogramme
der lebenden, transfektierten HEK293 Zellen (violett) wurden mit den Histogrammen der lebenden,
untransfektierten HEK293 Zellen (grün) überlagert. Positiv transfektierte Zellen wurden auf den Histogrammen mit
dem Gate M1 eingegrenzt und die Transfektionseffizienzen ermittelt. A: Dot Plot und Histogramm der Zellen, die mit
Calcium Phosphat transfektiert wurden. Es wurde eine Transfektionseffizienz von 46,88% erreicht. B: Dot Plot und
Histogramm jener Zellen, die mit dem Lipofectamine™ Reagent transfektiert wurden. Die Transfektionseffizienz
betrug 62,07%. C: Dot Plot und Histogramm jener HEK293 Zellen, die mit dem FuGENE®6 Reagenz transfektiert
wurden. Die Transfektionseffizienz betrug 19,92%.
2.3 Überprüfung der Plasmid DNA mittels Restriktionsfragmentanalyse
Da die Transfektion der HEK293 Zellen mit dem EGFP-N1 Vektor erfolgreich war und
sowohl die geeignete Zelldichte als auch die Transfektionsmethode für den zellulären
Assay gewählt wurden, sollten HEK293 Zellen im nächsten Schritt mit dem AT1R-EGFP
Vektor transfektiert werden. Im Zuge früherer Versuche von AFFiRiS AG wurde AT1R
cDNA bereits in einen EGFP-N1 Vektor kloniert. Durch das Einfügen der DNA an den N-
Terminus von EGFP, wurde die Expression eines C-terminalen Fusionsproteins
gewährleistet. Vor der Transfektion des AT1R-EGFP Vektors sollte nun die DNA mittels
Restriktionsfragmentanalyse überprüft werden.
Der EGFP-N1 Vektor und der AT1R-EGFP Vektor wurden mit den Restriktionsenzymen
BamHI und EcoRI einfach und doppelt verdaut. Die verdauten und unverdauten Vektoren
wurden auf ein 1%iges Agarosegel aufgetragen (vgl. Abb. 8). Die Proben der Restriktion
des EGFP-N1 Vektors wurden in die Taschen 1-4 und die Proben der Restriktion des
AT1R-EGFP Vektors in die Taschen 5-9 geladen. In die erste Tasche wurde der DNA
Marker geladen. Auf den Lanes 1-3 können in Abbildung 8 DNA Banden in einer Höhe
von ~5000kb gesehen werden. Da der EGFP-N1 Vektor eine Größe von 4.7kb aufweist,
deutete dies auf eine erfolgreiche Linearisierung des Vektors durch die
Restriktionsenzyme hin. Auf den Lanes 5 und 6 befinden sich DNA Banden des AT1R
12

Vektors in einer Höhe von ~6000kb. Da die AT1R cDNA eine Größe von 1077bp7 und der
EGFP-N1 Vektor eine Größe von 4.7kb aufweisen, ist die Größe dieser DNA Banden ein
Hinweis darauf, dass der AT1R-EGFP Vektor die AT1R cDNA enthält und der Vektor
durch die Restriktionsenzyme BamHI und EcoRI erfolgreich linearisiert wurde. Auf Lane 7
kann in Abbildung 8 der Doppelverdau des AT1R-EGFP Vektors gesehen werden Es
befinden sich, wie erwartet, zwei Banden auf dieser Lane: eine DNA Bande mit einer
Größe von ~4500kb und eine weitere DNA Bande mit einer Größe von ~1000kb. Dies ist
ein weiterer Hinweis darauf, dass der AT1R-EGFP Vektor die cDNA des Rezeptors enthält
und sie durch die Restriktionsenzyme BamHI und EcoRI erfolgreich herausgeschnitten
werden konnte.
Abb. 8: 1%iges Agarosegel zur Überprüfung der Plasmid DNA. Die Lanes 1-4 zeigen die DNA Banden des EGFP-N1
Vektors und die Lanes 5-8 jene des AT1R-EGFP Plasmids. Die Plasmide wurden mit dem Enzym BamHI (Lanes
1+5), mit dem Enzym EcoRI (Lanes 2+4) und mit beiden Enzymen (Lanes 3+7) verdaut. Lane 4 und Lane 8 zeigen
die Banden der unverdauten Vektoren. Auf Lane 0 wurde der DNA Marker aufgetragen.
2.4 Transfektionen unterschiedlicher DNA-Konstrukte
Für den zellulären Assay mussten HEK293 Zellen mit dem AT1R-EGFP Vektor und dem
NF-κB-luc Vektor kotransfektiert werden. Daher sollte in diesem Versuch die Effizienz
dieser Kotransfektion ermittelt werden. Da in vorhergegangenen Versuchen die
Transfektion des EGFP-N1 Vektors erfolgreich war und die Transfektionseffizienz bekannt
war, wurde diese Transfektion als Positivkontrolle durchgeführt. Als Negativkontrolle
diente die Transfektion des NF-κB-luc Konstrukts, da diese Transfektion zu keiner grünen
Fluoreszenz führen sollte. Außerdem wurden HEK293 Zellen mit den Vektoren NF-κB-luc
und EGFP-N1 kotransfektiert.
Jene Zellen, die mit dem NF-κB-luc Vektor transfektiert wurden, zeigten bei der FACS
Analyse erwartungsgemäß keine grüne Fluoreszenz, da dieser Vektor kein EGFP enthält
(vgl. Abb. 9A). Die Transfektion des EGFP-N1 Vektors ergab erneut mit 66,98% eine sehr
13
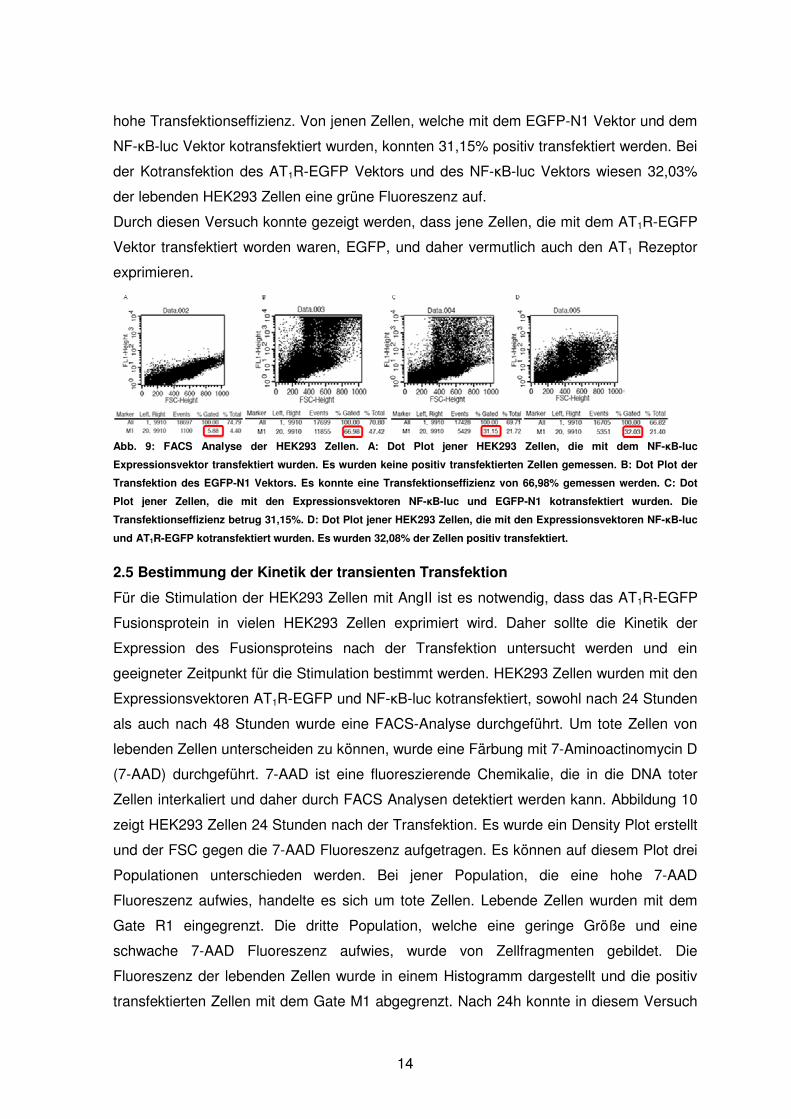
hohe Transfektionseffizienz. Von jenen Zellen, welche mit dem EGFP-N1 Vektor und dem
NF-κB-luc Vektor kotransfektiert wurden, konnten 31,15% positiv transfektiert werden. Bei
der Kotransfektion des AT1R-EGFP Vektors und des NF-κB-luc Vektors wiesen 32,03%
der lebenden HEK293 Zellen eine grüne Fluoreszenz auf.
Durch diesen Versuch konnte gezeigt werden, dass jene Zellen, die mit dem AT1R-EGFP
Vektor transfektiert worden waren, EGFP, und daher vermutlich auch den AT1 Rezeptor
exprimieren.
Abb. 9: FACS Analyse der HEK293 Zellen. A: Dot Plot jener HEK293 Zellen, die mit dem NF-κB-luc
Expressionsvektor transfektiert wurden. Es wurden keine positiv transfektierten Zellen gemessen. B: Dot Plot der
Transfektion des EGFP-N1 Vektors. Es konnte eine Transfektionseffizienz von 66,98% gemessen werden. C: Dot
Plot jener Zellen, die mit den Expressionsvektoren NF-κB-luc und EGFP-N1 kotransfektiert wurden. Die
Transfektionseffizienz betrug 31,15%. D: Dot Plot jener HEK293 Zellen, die mit den Expressionsvektoren NF-κB-luc
und AT1R-EGFP kotransfektiert wurden. Es wurden 32,08% der Zellen positiv transfektiert.
2.5 Bestimmung der Kinetik der transienten Transfektion
Für die Stimulation der HEK293 Zellen mit AngII ist es notwendig, dass das AT1R-EGFP
Fusionsprotein in vielen HEK293 Zellen exprimiert wird. Daher sollte die Kinetik der
Expression des Fusionsproteins nach der Transfektion untersucht werden und ein
geeigneter Zeitpunkt für die Stimulation bestimmt werden. HEK293 Zellen wurden mit den
Expressionsvektoren AT1R-EGFP und NF-κB-luc kotransfektiert, sowohl nach 24 Stunden
als auch nach 48 Stunden wurde eine FACS-Analyse durchgeführt. Um tote Zellen von
lebenden Zellen unterscheiden zu können, wurde eine Färbung mit 7-Aminoactinomycin D
(7-AAD) durchgeführt. 7-AAD ist eine fluoreszierende Chemikalie, die in die DNA toter
Zellen interkaliert und daher durch FACS Analysen detektiert werden kann. Abbildung 10
zeigt HEK293 Zellen 24 Stunden nach der Transfektion. Es wurde ein Density Plot erstellt
und der FSC gegen die 7-AAD Fluoreszenz aufgetragen. Es können auf diesem Plot drei
Populationen unterschieden werden. Bei jener Population, die eine hohe 7-AAD
Fluoreszenz aufwies, handelte es sich um tote Zellen. Lebende Zellen wurden mit dem
Gate R1 eingegrenzt. Die dritte Population, welche eine geringe Größe und eine
schwache 7-AAD Fluoreszenz aufwies, wurde von Zellfragmenten gebildet. Die
Fluoreszenz der lebenden Zellen wurde in einem Histogramm dargestellt und die positiv
transfektierten Zellen mit dem Gate M1 abgegrenzt. Nach 24h konnte in diesem Versuch
14

eine Transfektionseffizienz von 17,37% gemessen werden. Die Anzahl der toten Zellen
belief sich auf ~20% (vgl. Abb. 10C).
Abb. 10: Kinetikstudie der AT1R-EGFP Expression durch eine FACS Analyse nach 24h. A: Es wurde eine 7-AAD
Färbung durchgeführt und ein Density Plot erstellt. Das Gate R1 schließt die lebenden Zellen ein. B: Das
Histogramm beinhaltet ausschließlich die Zellen des Gates R1. Durch das Gate M1 wurden die positiv
transfektierten Zellen eingegrenzt. C: Die Statistik des Histogramms zeigt, dass die Transfektionseffizienz 17,37%
betrug.
Nach 48h wurden die FACS Analyse und die 7-AAD Färbung wiederholt. Es konnte
bereits eine Abnahme der Transfektionseffizienz und eine Zunahme der Anzahl der toten
Zellen gemessen werden. Die Transfektionseffizienz betrug 14,77% und die Anzahl der
toten Zellen stieg von ~20% auf ~40% (vgl. Abb. 11 C).
Abb. 11: Kinetikstudie der AT1R-EGFP Expression durch eine FACS Analyse nach 48h. A: Es wurde eine 7-AAD
Färbung durchgeführt und ein Density Plot erstellt. Auf diesen Density Plot wurde ein Gate R1 gesetzt, das die
lebenden Zellen einschließt. B: Das Histogramm zeigt nur jene Zellen, die durch das Gate R1 eingeschlossen
wurden. Ein weiteres Gate M1 wurde gesetzt, das die positiv transfektierten Zellen eingrenzt. C: Die Statistik zeigt,
dass di e Transfektionseffizienz 14,77% betrug.
Diese Daten zeigten, dass nach 48 Stunden die Expression des AT1R-EGFP
Fusionsproteins bereits abnimmt und eine Vielzahl von HEK293 Zellen bereits tot ist.
Daher wurde beschlossen die Stimulation für den zellulären Assay nach 24 Stunden
durchzuführen.
2.6 Lokalisierung des AT1 Rezeptors mittels Fluoreszenzmikroskopie
In diesem Versuch sollte mit einem Fluoreszenzmikroskop überprüft werden, ob der AT1
Rezeptor nach der Transfektion des AT1R-EGFP Expressionsvektors an der Oberfläche
von HEK293 Zellen exprimiert wird.
Es wurden zunächst die untransfektierten HEK293 Zellen mikroskopiert. Dabei konnte
gesehen werden, dass auch diese HEK293 Zellen eine schwache Eigenfluoreszenz
aufweisen. Diese konnte hauptsächlich intrazellulär in granulären Strukturen erkannt
werden (vgl. Abb. 12).
15
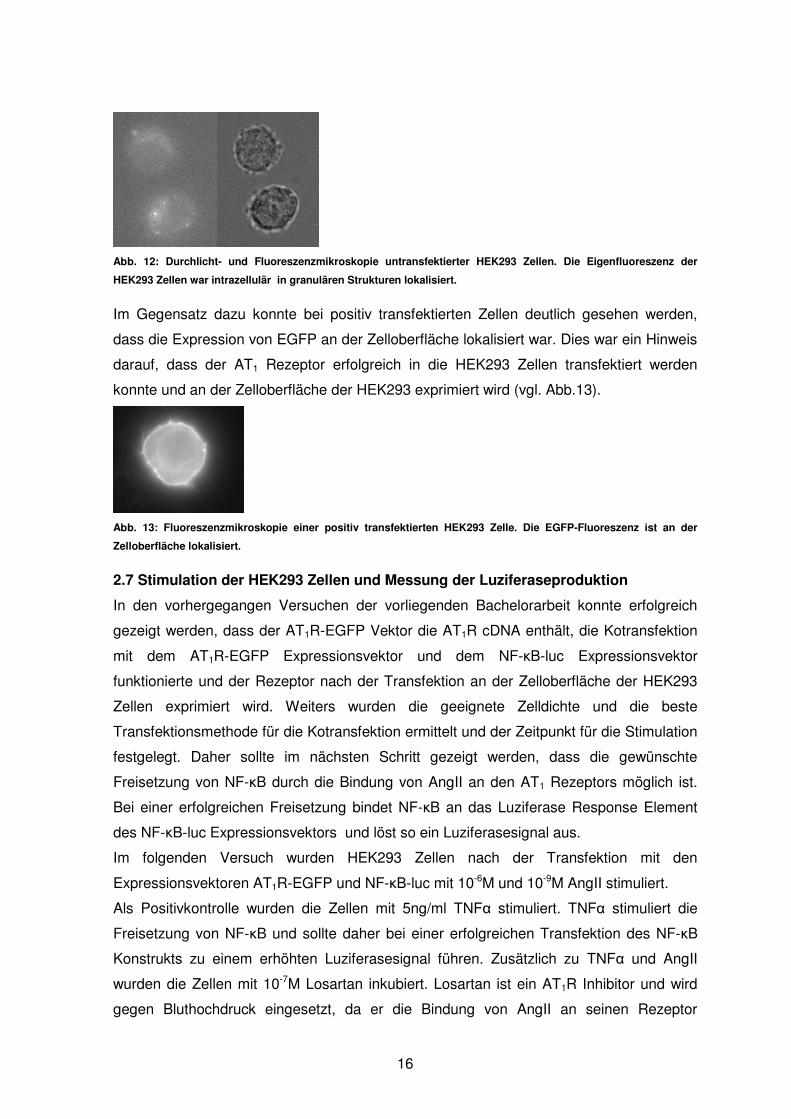
Abb. 12: Durchlicht- und Fluoreszenzmikroskopie untransfektierter HEK293 Zellen. Die Eigenfluoreszenz der
HEK293 Ze llen war intrazellulär in granulären Strukturen lokalisiert.
Im Gegensatz dazu konnte bei positiv transfektierten Zellen deutlich gesehen werden,
dass die Expression von EGFP an der Zelloberfläche lokalisiert war. Dies war ein Hinweis
darauf, dass der AT1 Rezeptor erfolgreich in die HEK293 Zellen transfektiert werden
konnte und an der Zelloberfläche der HEK293 exprimiert wird (vgl. Abb.13).
Abb. 13: Fluoreszenzmikroskopie einer positiv transfektierten HEK293 Zelle. Die EGFP-Fluoreszenz ist an der
Zelloberfläche lokalisiert.
2.7 Stimulation der HEK293 Zellen und Messung der Luziferaseproduktion
In den vorhergegangen Versuchen der vorliegenden Bachelorarbeit konnte erfolgreich
gezeigt werden, dass der AT1R-EGFP Vektor die AT1R cDNA enthält, die Kotransfektion
mit dem AT1R-EGFP Expressionsvektor und dem NF-κB-luc Expressionsvektor
funktionierte und der Rezeptor nach der Transfektion an der Zelloberfläche der HEK293
Zellen exprimiert wird. Weiters wurden die geeignete Zelldichte und die beste
Transfektionsmethode für die Kotransfektion ermittelt und der Zeitpunkt für die Stimulation
festgelegt. Daher sollte im nächsten Schritt gezeigt werden, dass die gewünschte
Freisetzung von NF-κB durch die Bindung von AngII an den AT1 Rezeptors möglich ist.
Bei einer erfolgreichen Freisetzung bindet NF-κB an das Luziferase Response Element
des NF-κB-luc Expressionsvektors und löst so ein Luziferasesignal aus.
Im folgenden Versuch wurden HEK293 Zellen nach der Transfektion mit den
Expressionsvektoren AT1R-EGFP und NF-κB-luc mit 10-6M und 10-9M AngII stimuliert.
Als Positivkontrolle wurden die Zellen mit 5ng/ml TNFα stimuliert. TNFα stimuliert die
Freisetzung von NF-κB und sollte daher bei einer erfolgreichen Transfektion des NF-κB
Konstrukts zu einem erhöhten Luziferasesignal führen. Zusätzlich zu TNFα und AngII
wurden die Zellen mit 10-7M Losartan inkubiert. Losartan ist ein AT1R Inhibitor und wird
gegen Bluthochdruck eingesetzt, da er die Bindung von AngII an seinen Rezeptor
16

verhindert.4 Losartan sollte demnach das durch AngII ausgelöste Luziferase Signal
hemmen, jedoch keinen Einfluss auf das durch TNFα ausgelöste Signal haben.
In diesem Versuch führte die Stimulation der HEK293 Zellen mit TNFα zu einem sehr
starken Anstieg der Lumineszenz. Allerdings wurde das Signal durch die Zugabe von
10-7M Losartan geschwächt (vgl. Abb. 14).
Abb. 14: Ergebnisse des Luziferase Assays. Die durchgeführten Stimulationen wurden auf der x-Achse und die
Lumineszenz auf der y-Achse aufgetragen. Das durch TNFα ausgelöste Signal erreichte den maximalen Messwert,
wurde jedoch durch Losartan geschwächt. AngII konnte hingegen nur ein schwaches Signal auslösen, welches
jedoch durch Losartan stark he rabgesetzt werden konnte.
Die Stimulationen mit 10-9M und 10-6M AngII führte im Vergleich zu TNFα nur zu einem
geringen Anstieg der Lumineszenz, deshalb wurden in Abbildung 15 die Ergebnisse der
Stimulationen mit AngII getrennt dargestellt. Es konnte mit 10-9M AngII ein höheres Signal
induziert werden als mit 10-6M AngII. Durch die Zugabe von Losartan konnte das durch
10-6M AngII-induzierte Signal um den Faktor fünf geschwächt werden.
Abb. 15: Ergebnisse der Stimulation von HEK293 Zellen mit AngII und Losartan. Das Signal von 10-9M AngII ist
deutlich höher als jenes von 10-6M AngII. Durch die Zugabe von 10-7M Losartan konnte das AngII Signal um den
Faktor fünf geschwächt werden.
In einem weiteren Versuch wurden nach der Kotransfektion mit den Expressionsvektoren
AT1R-EGFP und NF-κB-luc, 10-6M AngII, TNFα und Losartan zu den HEK293 Zellen
hinzugegeben. Dieser Luziferase Assay zeigte allgemein sehr hohe Lumineszenzsignale,
was durch ein starkes Hintergrundsignal erklärbar ist. Trotzdem konnte sowohl bei der
Stimulation mit TNFα als auch bei der Stimulation mit 10-6M AngII eine deutliche Zunahme
der Luziferaseproduktion gemessen werden. Das durch 10-6M AngII-induzierte Signal
konnte auch bei diesem Versuch mit 10-7M Losartan stark herabgesetzt werden. Es
17

konnte in mehreren Assays beobachtet werden, dass Zellen, die mit Losartan und AngII
stimuliert wurden, sogar ein schwächeres Lumineszenzsignal aufwiesen als unstimulierte
Zellen.
Abb. 16: Lumineszenzsignale unstimulierter und stimulierter HEK293 Zellen. Die Zellen wurden mit 10-6M AngII,
TNF , 10-7α M Losartan stimuliert. Das S ignal der AngII Stimuation wurde durch Losartan stark g eschwächt.
2.8 Testung monoklonaler Antikörper
Da die Stimulation mit AngII ein deutliches Luziferase Signal induzierte und das Signal
durch Losartan gehemmt werden konnte, sollte nun die inhibitorische Fähigkeit von
monoklonalen Antikörpern getestet werden. Es wurden HEK293 wie schon in den
Versuchen zuvor, mit den Vektoren AT1R-EGFP und NF-κB-luc kotransfektiert und mit
10-6M AngII stimuliert. Durch die Zugabe eines AngII spezifischen monoklonalen
Antikörpers (mAb 32-4-4) sollte gezeigt werden, dass das AngII-induzierte Signal durch
den Antikörper geblockt werden kann. Als Isotyp Kontrolle wurde ein irrelevanter,
monoklonaler Antiköper verwendet (mAb 151). Es konnte gemessen werden, dass das
AngII-induzierte Signal durch den Kontrollantikörper nicht beeinträchtigt wurde, es
hingegen durch den AngII spezifischen Antikörper erfolgreich herabgesetzt werden
konnte. Dies zeigte, dass der monoklonale Antikörper 32-4-4 das AngII-induzierte Signal
spezifisch inhibieren konnte.
Abb. 17: Überprüfung der inhibitorischen Fähigkeit v on monoklonalen Antikörpern. Die S timulation mit AngII führte
zu einer signifikanten Steigung des Lumineszenzsignals. Der monoklonale Kontrollantiköper hatte keinen Einfluss
auf di eses Signal. Der AngII spezifische monoklonale Antiköper hingegen konnte d ieses Signal herabsetzen.
18

3. Diskussion
Im Zuge der vorliegenden Bachelorarbeit sollte ein zellulärer in vitro Assay in HEK293
Zellen etabliert werden, mit dem es möglich ist die Aktivität von AngII durch die
Aktivierung des Transkriptionsfaktors NF-κB zu messen.
Da HEK293 Zellen den AT1 Rezeptor nicht endogen exprimieren, wurden sie mit einem
AT1R-EGFP Expressionsvektor transient transfektiert. EGFP ist ein fluoreszierendes
Protein, das nach seiner Aktivierung eine grüne Fluoreszenz aufweist, und daher leicht
detektiert werden kann. Diese Eigenschaft ermöglicht zum einen die Bestimmung der
Transfektionseffizienz durch FACS Analysen und zum anderen die Lokalisation des AT1
Rezeptors nach seiner Expression in der Zelle mittels Fluoreszenzmikroskopie. Die
Funktionalität eines AT1R-EGFP Fusionsproteins in HEK293 Zellen wurde bereits 2002
durch Versuche von Hunyady bestätigt.8
Um zu überprüfen, ob der AT1R-EGFP Expressionsvektor die cDNA des Rezeptors
tatsächlich enthält, wurde eine Restriktionsfragmentanalyse durchgeführt. Diese zeigte,
dass der AT1R-EGFP Expressionsvektor sowohl mit dem Restriktionsenzym BamHI als
auch mit dem Restriktionsenzym EcoRI linearisiert werden konnte. Der Doppelverdau des
Vektors führte erwartungsgemäß zu zwei DNA Banden. Allerdings wiesen diese DNA
Banden Größen von ~4500bp und ~900-1000bp, anstatt 4700bp und 1077bp auf. Eine
Erklärung für diese Abweichungen könnte sein, dass das Gel ungleichmäßig lief und
daher die DNA Fragmente unterschiedlich schnell aufgetrennt wurden. In Anbetracht
dessen kann auf Grund der Ergebnisse angenommen werden, dass der AT1R-EGFP
Expressionsvektor die AT1R cDNA enthält und daher sowohl EGFP als auch der Rezeptor
in den Zellen exprimiert werden können.
Die Ergebnisse dieser Restriktionsfragmentanalyse konnten in dieser Arbeit durch FACS
Analysen bestätigt werden. Es konnte gezeigt werden, dass HEK293 Zellen, die mit den
Expressionsvektoren AT1R-EGFP und NF-κB-luc transient kotransfektiert wurden, eine
grüne Fluoreszenz aufwiesen. Dies zeigte, dass die Zellen erfolgreich mit dem AT1R
EGFP Vektor transfektiert werden konnten und EGFP exprimierten. Da die AT1R-cDNA an
den N-Terminus des EGFP-Gens angefügt wurde, kann angenommen werden, dass auch
der Rezeptor erfolgreich exprimiert werden konnte.
Allerdings wiesen Zellen, die mit dem AT1R-EGFP Konstrukt transfektiert worden waren,
eine geringere Fluoreszenzintensität auf als jene Zellen, die mit dem EGFP-N1 Vektor
alleine transfektiert wurden. Die Tatsache, dass anstatt EGFP nun das gesamte
Fusionsprotein exprimiert werden musste, könnte diese Intensitätsabnahme erklären.
Eine weitere Bestätigung für die erfolgreiche Expression des Rezeptors erbrachte die
Fluoreszenzmikroskopie. Es konnte gezeigt werden, dass die Expression von EGFP in
19

positiv transfektierten Zellen an der Zelloberfläche lokalisiert war, wo auch die Expression
des Rezeptors erwartet wurde.
Um zu überprüfen, ob der Rezeptor auch funktionell aktiv ist, wurden kotransfektierte
HEK293 Zellen mit unterschiedlichen AngII Konzentrationen stimuliert. Durch diese
Stimulation konnte in einigen Assays eine signifikante Erhöhung der Luziferaseproduktion
erzielt werden. Um die Spezifität dieser Signalerhöhung zu überprüfen wurden die Zellen
zusätzlich zu AngII mit Losartan inkubiert. Losartan ist ein AT1R Inhibitor, der die Bindung
von AngII an seinen Rezeptor verhindert.4 Das AngII-induzierte Signal konnte durch
Losartan gänzlich unterdrückt werden. Diese Tatsache zeigte, dass das Signal spezifisch
durch AngII ausgelöst wurde und der AT1 Rezeptor daher funktionell aktiv sein musste.
Eine weitere Bestätigung für die Funktionalität des Assays und die Spezifität des AngII
Signales erbrachte die Testung von monoklonalen Antikörpern. Es konnte gezeigt
werden, dass ein AngII spezifischer monoklonaler Antikörper das AngII-induzierte
Luziferasesignal schwächen konnte, während ein AngII unspezifischer Kontrollantikörper
diesen Effekt nicht auslöste.
Es konnte also gezeigt werden, dass HEK293 Zellen, die mit dem AT1R-EGFP
Expressionsvektor transfektiert wurden, sowohl EGFP als auch den AT1 Rezeptor
exprimierten und der Rezeptor in den Zellen funktionell aktiv war. Weiters konnte die
Funktionalität des Assays durch die erfolgreiche Inhibierung des AngII-induzierten Signals
mit einem AngII spezifischen, monoklonalen Antikörper, bestätigt werden.
Allerdings konnten auch einige unsichere Faktoren im Assay ausgemacht werden. Es
wurde im Zuge von Versuchen festgestellt, dass zwischen unterschiedlichen Wells große
Schwankungen in den Transfektionseffizienzen auftraten. Da in den ersten Versuchen
jeweils ein Well pro Stimulus verwendet wurde, konnten die Ergebnisse auf Grund
unterschiedlicher Transfektionseffizienzen oftmals nicht verglichen werden. Daher
müssen in der vorliegenden Bachelorabeit die Ergebnisse des ersten Luziferase Assays
mit Vorsicht betrachtet werden. In weiteren Versuchen wurden, um diesem Problem
entgegenzuwirken, mehrere Wells pro Stimulus verwendet und der Mittelwert für die
Auswertungen berechnet. Um für die Stimulation gleichwertige Bedingungen zu schaffen,
wurde nach einer Methode gesucht, mit der es möglich ist, gleiche Transfektions
effizienzen in unterschiedlichen Wells zu erreichen. Hierfür wurden Zellen für die
Transfektion gepoolt und anschließend auf Wellplatten aufgeteilt. Durch FACS Analysen
konnte gemessen werden, dass mit dieser Methode gleiche Transfektionseffizienzen von
Well zu Well geschaffen werden konnten.
Doch auch die Stimulation mit AngII per se stellt in diesem Assay einen unsicheren Faktor
dar. Während TNFα beständig zu sehr hohen Signalanstiegen führte, führte die
Stimulation mit AngII oft nur zu einer schwachen Steigerung der Lumineszenzsignale. Es
20

wurde zunächst vermutet, dass das NF-κB-luc Konstrukt nicht erfolgreich transfektiert
werden konnte, da TNFα jedoch durchwegs zu starken Luziferasesignalen führte, konnte
dies ausgeschlossen werden. Dies könnte also bedeuten, dass die Aktivierung von NF-κB
durch AngII allgemein nicht gut funktionierte und der Transkriptionsfaktor daher kein
optimales Kontrollelement in diesem Assay darstellt.
Ruiz-Ortega et al. bestimmten den Effekt von AngII auf die Aktivität von NF-κB in glatten
Gefäßmuskelzellen (VSMC) von Ratten. Sie transfektierten die Zellen transient mit einem
NF-kB-luc Konstrukt und stimulierten sie mit AngII. Nach der Stimulation maßen sie, wie
auch in der vorliegenden Arbeit, die Luziferaseproduktion. Sie konnten zeigen, dass die
Stimulation der Zellen mit 10-7M AngII zu einer 6-fachen Steigerung der
Luziferaseproduktion führte.9 In der vorliegenden Bachelorarbeit konnte hingegen meist
nur eine 2-fache Steigerung der Luziferase Aktivität erzielt werden. Ein Grund für diese
Ergebnisse könnte die Tatsache sein, dass glatte Gefäßmuskelzellen, im Gegensatz zu
HEK293 Zellen, den AT1 Rezeptor endogen exprimieren und daher auch mit AngII
erfolgreicher stimuliert werden können.
In einigen Assays führte die Zugabe von Losartan zu einer Schwächung des TNFα
induzierten Luziferasesignals. Da Losartan in den ersten Versuchen vier Stunden vor den
restlichen Stimuli zu den Zellen hinzugefügt wurde, wurde vermutet, dass Losartan durch
diese Präinkubation toxisch auf die Zellen wirkte und dadurch das Signal von TNFα
herabgesetzt wurde. Dies würde jedoch bedeuten, dass auch die Blockade des AngII
induzierten Signals durch die toxische Wirkung und nicht auf Grund einer spezifischen
Inhibierung herabgesetzt wurde. Da jedoch das Signal von TNFα durch Losartan nur
geschwächt und das Signal von AngII beinahe gänzlich blockiert wurde, wurde auch in
diesen Versuchen die Blockade des AngII-induzierten Signals durch Losartan als
spezifisch angesehen.
Zusammenfassend konnte in dieser Bachelorarbeit gezeigt werden, dass durch den
zellulären Assay spezifische AngII Signale gemessen und durch monoklonale Antikörper
geschwächt werden können. Da dieser Assay in diesem Stadium jedoch noch sehr
instabil ist, kann er für die Testung VARIOTOP®-induzierter Antikörper nicht
herangezogen werden. Eine Möglichkeit diesen Assay stabiler zu gestalten, wäre die
Verwendung einer stabil transfektierten Zelllinie. In zukünftigen Versuchen muss
außerdem entschieden werden, ob die Aktivierung von NF-κB durch AngII tatsächlich ein
geeignetes Kontrollelement darstellt.
21

4. Literaturverzeichnis
1 Pandey R. et al. (2009): Vaccine for hypertension: Modulating the rennin-angiotensin
system. Journal of Cardiology 134,160-168
2 Maurer P. et al. (2009): Immunization against angiotensins for the treatment of
hypertension. Clinical Immunology 134, 89-95
3 de Gasparo M. et al. (2000): International Union of Pharmacology. XXIII. The
Angiotensin II Receptors: Pharmacological Reviews 52, 415-472
4 Zaman M. A. et al. (2002): Drugs targeting the renin-angiotensin-aldosterone system.
Nature Reviews Drug discovery 1, 621-636
5 Wenzel U. O. et al. (2009): The angiotensin II type 2 receptor in renal disease. Journal of
the Renin-Angiotensin-Aldosterone System 11, 37-41
6 Guo R. et al. (2006): Angiotensin II induces NF-kB activation in HUVEC via the
p38MAPK pathway. PEPTIDES 27, 3269-3275
7 Schussek S. (2009): Evaluation of candidate peptides for the immunisation against
Angiotensin II. Diplomarbeit, Universität Wien.
8 Hunyady L. et al. (2002): Differential PI 3-kinase dependence of early and late phases of
recycling of the internalized AT1 angiotensin receptor. Journal of Cell Biology 157, 1211
1222
9 Ruiz-Ortega M. et al. (2000): Angiotensin II Activates Nuclear Transcription Factor κB
Through AT1 and AT2 in Vascular Smooth Muscle Cells. Circulation Research 86, 1266
1272
22

5. Eigenständigkeitserklärung
23

6. Anhang
6.1 Materialien und Methoden
Zellkultur
HEK293 Zellen werden in Dulbecco`s modified Eagles medium (DMEM) + GlutaMAX™
mit 10% fetalem Kälberserum (FCS) und Penicillin/Streptomycin (P/S) kultiviert. Die Zellen
werden zweimal in der Woche 1:20 gesplittet.
Für die Zellzählung werden 10µl Zellsuspension mit 140µl 1xPBS und 50µl 0.4%
Trypanblau (GIBCO) versetzt. Mit Hilfe einer Neubauer Zählkammer werden die Zellen
unter einem Lichtmikroskop gezählt.
Die Zellanzahl pro Milliliter wird durch folgende Rechnung berechnet:
Gezählte Zellen x Verdünnungsfaktor x 104
Anzahl der Quadrate in Zählkammer
Transfektion der HEK293 Zellen
Calcium Phosphat Transfektion
0,25-1x106 Zellen/ml werden einen Tag vor der Transfektion in DMEM mit 10% FCS und
P/S in einer Petrischale (16ml), oder in einer 6-Well Platte (2ml/Well) ausgesät. Am
folgenden Tag wird das Medium entfernt und FCS freies DMEM mit P/S hinzugefügt. Für
die Transfektion wird zunächst die Plasmid DNA in der entsprechenden Menge H2O und
CaCl2 verdünnt. Zuletzt wird der HeBS- Puffer tropfenweise und unter leichtem Schütteln
hinzugefügt.
Mengenangaben für ein Well einer 6-well Platte (zu je 2ml)
2µg AT1R-EGFP Plasmid DNA
2µg NF-κB-luc Plasmid DNA
9µl 2M CaCl2
71µl HeBS Puffer
61µl H2O
Der Ansatz wird tropfenweise zu den Zellen pipettiert. Nach 4 bis 5 Stunden wird die
Transfektionslösung entfernt und frisches Medium (DMEM mit 10% FCS und P/S)
hinzugefügt.
24

Transfektion mit FuGENE®6 Transfection Reagent (Roche)
Es werden drei Ansätze mit drei unterschiedlichen FuGENE®6 Reagenz Konzentrationen
durchgeführt, sodass das Verhältnis zwischen Plasmid DNA und Reagenz 1:6,1:3 und 2:3
beträgt. Zunächst wird FCS und P/S freies DMEM in der entsprechenden Menge in
Eppendorf Tubes vorgelegt und das FuGENE®6 Reagenz hinzupipettiert. Der Ansatz wird
kurz gevortext und für 5 Minuten bei Raumtemperatur inkubiert. Zuletzt wird die Plasmid
DNA in dem entsprechenden Verhältnis hinzugefügt. Der gesamte Ansatz wird gevortext
und für 15 Minuten bei Raumtemperatur inkubiert. Anschließend wird der Ansatz
tropfenweise zu den Zellen hinzugefügt. Nach 4 bis 5 Stunden wird das Medium
abgenommen und DMEM mit 10%FCS und P/S hinzugefügt.
(Siehe Protokoll „FuGENE®6 Transfection Reagent“ von Roche)
Transfektion mit Lipofectamine™ Reagent und PLUS® Reagent (Invitrogen)
1µg EGFP-N1 Plasmid DNA wird in 100µl FCS freiem DMEM mit P/S verdünnt. Das Plus®
Reagenz wird gevortext und 10µl zu der verdünnten DNA hinzugefügt. Der Ansatz wird für
15 Minuten bei Raumtemperatur inkubiert.
10µl Lipofectamine™ Reagenz werden ebenfalls in 100µl FCS freiem DMEM mit P/S
verdünnt und nach der Inkubationszeit zu dem restlichen Ansatz hinzugefügt und erneut
für 15 Minuten bei Raumtemperatur inkubiert.
Anschließend wird der Ansatz tropfenweise zu den Zellen hinzugefügt. Nach 4 bis 5
Stunden wird das Medium abgenommen und DMEM mit 10%FCS und P/S hinzugefügt.
(siehe Protokoll „Lipofectamine™ Reagent“ von Invitrogen)
Stimulation der transfektierten HEK293 Zellen
Stimulation mit AngII, TNFα und Inhibierung mit Losartan
Die Stimulation der HEK293 Zellen wird 24h Stunden nach der Transfektion in einer 6
Well Platte oder einer 24-Well Platte durchgeführt. Vor Zugabe der Stimuli wird das
Medium entfernt und FCS freies DMEM hinzugefügt. Die Zellen werden mit 10-7M
Losartan (Sigma-Aldrich), 10-6M und 10-9M Angiotensin II und 5ng/ml TNFα (R&D
Systems) stimuliert. Losartan wird 2 bis 4 Stunden vor der Stimulation mit AngII und TNFα
zu den Zellen hinzugefügt. Nach der Inkubationszeit werden die restlichen Stimuli
hinzugefügt und die Mediumsschale vorsichtig geschwenkt um die Stimuli zu verteilen.
Stimulation mit AngII und Testung monoklonaler Antikörpern
10-6M AngII und 0,6x10-6M AngII-spezifischer Antikörper (mAb 32-4-4) bzw. 0,4x10-6M
Kontrollantikörper (mAb 151) werden für 30 Minuten in 2ml FCS freiem DMEM in
25

Eppendorf Tubes präinkubiert. Anschließend wird das Medium von den Hek293 Zellen
abgenommen und der AngII-Antikörper-Mix zu den Zellen hinzugefügt.
Luziferase Assay
24 Stunden nach der Stimulation werden die Zellen mit 300µl des Lysis Buffers (Super
Light Luciferase Reporter Gene Assay Kit, BioAssay Systems; Gentaur) abgelöst. 200µl
der abgelösten Zellen werden entnommen und auf zwei Wells einer 96-Well Platte (black
VIEW plate PerkinElmar) zu je 100µl aufgeteilt. Anschließend wird das Luziferase Signal
mit dem TECAN GENios Reader (Luminometer) und dem Programm luminescence.mth
gemessen. Der Emissionsfilter und der Anregungsfilter werden hierfür zuvor entfernt.
(Integrationszeit: 500msec)
FACS (Fluorescence Activated Cell Sorting) Analyse
Die FACS Analyse ermöglicht die Zählung von Zellen und die Messung ihrer Größe, ihrer
Granularität und ihrer Fluoreszenz. Die Zellen werden einzeln angeordnet und von einem
Laser bestrahlt. Trifft ein Laserstrahl auf eine Zelle, wird dieser, abhängig von der Größe
und der Oberflächenbeschaffenheit jener Zelle, unterschiedlich stark gestreut. Durch
diese Streuung können Größe und Granularität bestimmt werden. Dabei misst der Foward
Scatter die Größe der Zellen, während der Side Scatter die Granularität der Zellen angibt.
Außerdem können Fluoreszenzen detektiert werden und so Zellen selektiert werden. Die
FACS Analysen werden mit dem FACScan Flow Cytometer 65 von Becton Dickinson
GmbH und dem Programm CellQuest Pro durchgeführt. Für die Messung werden 2x105
4x105 Zellen entnommen, für 3 bis 4 Minuten bei 1000rpm abzentrifugiert und der
Überstand abgenommen. Das Pellet wird anschließend in 400µl eiskaltem FACS Puffer
resuspendiert.
7-AAD Färbung
Kurz vor der FACS-Analyse wird 7-AAD (Stocklösung 1mg/ml) in einem Verhältnis von
1:1000 zu den Messproben hinzugefügt und kurz gevortext. Bei 7-AAD handelt es sich um
7-Aminoactinomycin D. Diese fluoreszierende Chemikalie kann in die DNA von toten
Zellen interkalieren, da deren Zellmamembran löchrig und dadurch durchlässig werden.
Restriktionsfragmentanalyse
1µg DNA wird mit 1µg Enzym (BamHI, EcoRI von Fermentas), 16µl ddH2O und 2µl
Reaktionspuffer (1x BamHI Buffer von Fermentas) für 3 bis 5 Stunden bei 37°C inkubiert.
Anschließend werden die Verdaue auf ein 1%iges Agarosegel geladen (~45 Minuten,
100V).
26

Agarosegelelektrophorese
Es wird 1%iges Agarosegel in 1xTAE in der Mikrowelle erhitzt und 10µl (1:10000) Sybr
Safe DNA Stain (Invitrogen) hinzugefügt. Als DNA Marker wird der GeneRuler™ 1kB DNA
Ladder (Fermentas) verwendet. Das Gel wird in 1xTAE für ungefähr 45 Minuten bei 100V
laufen gelassen und die Banden anschließend unter UV Licht im Transluminator M-26 (bio
Doc-It System) angesehen. Als Ladepuffer wurde ein Puffer aus Xylencyanol und Orange
Green (Sigma) verwendet.
Durchlicht- und Fluoreszenzmikroskopie
HEK293 Zellen werden mit 1xPBS von der Zellkulturschale abgelöst und 600µl in ein
Eppendorf Tube überführt. Die Zellen werden bei 1000rpm für 4 Minuten abzentrifugiert.
Der Überstand wird verworfen und das Pellet in eiskaltem 1xPBS resuspendiert. Die
Zellen werden tropfenweise auf einen Objektträger überführt und für 2 bis 3 Minuten
inkubiert. Anschließend wurden die Zellen mikroskopiert.
Allgemeine Puffer
1xPBS
137 mM NaCl
2,7 mM KCl
4,3 mM Na2HPO4
1,47 mM KH2PO4
pH 7,4-7,6
Ladungs Puffer für DNA Gel
10 mM Tris-HCl (pH 7,6)
0,15% Orange Green (Sigma O3756)
0,03% Xylencyanol FF (Sigma X4126)
60% Glycerol (Roth 7530.1)
60 mM EDTA pH 8,0
27

HeBS Puffer
280mM NaCl
1,5mM Na2HPO4
50mM Hepes (Sigma H-7006)
FACS Puffer
1xPBS
1%BSA
0,1%NaN3
28

6.2 Abkürzungsverzeichnis
7-AAD 7-Aminoactinomycin D
ACE Angiotensin Converting Enzyme
AngI Angiotensin I
AngII Angiotensin II
AngIII Angiotensin III
AngIV Angiotensin IV
As Aminosäure
AT1R Angiotensin Typ 1 Rezeptor
AT2R Angiotensin Typ 2 Rezeptor
DAG Diacylglycerol
DMEM Dulbecco`s modified Eagles medium
DNA Desoxyribonukleinsäure
EGFP Enhanced Green Fluorescent Protein
FACS Fluorescence Activated Cell Sorting
FCS Fetales Kälber Serum
FL1 Fluoreszenz 1
FSC Forward Scatter
HEK293 Zellen Human Embryonic Kidney cells
Hepes 2-(4-(2-Hydroxyethyl)-1-piperazinyl)
ethansulfonsäure
I-κB Inhibitor von κB
IP3 Inositol-1,4,5-triphosphat
MCS Multiple Cloning Site
NF-κB Nukleärer Faktor κB
NF-κB-luc NF-κB Response-Element Luziferase
Reporter Gen Expressionsvektor
NO Stickstoffmonoxid
PBS Phosphate Buffer Saline
PLC Phospholipase C
P/S Penicillin/Streptomycin
RAS Renin-Angiotensin System
SSC Side Scatter
TAE Tris Acetat
Ethylendiamintetraessigsäure
TNFα Tumornekrosefaktor α
VSMC glatte Gefäßmuskelzellen
29

6.3 Abbildungsverzeichnis
Abbildung 1: Eigene Graphik
Abbildung 2: Eigene Graphik
Abbildung 3: Schussek, S (2009): Evaluation of candidate peptides for the immunisation
against angiotensin II. Diplomarbeit, Universität Wien. S. 45
Abbildung 4: Eigene Graphik
Abbildung 5: siehe oben
Abbildung 6: siehe oben
Abbildung 7: siehe oben
Abbildung 8: siehe oben
Abbildung 9: siehe oben
Abbildung 10: siehe oben
Abbildung 11: siehe oben
Abbildung 12: siehe oben
Abbildung 13: siehe oben
Abbildung 14: siehe oben
Abbildung 15: siehe oben
Abbildung 16: siehe oben
Abbildung 17: siehe oben
30

6.4 Protokolle
Online unter: http://www.roche-applied-science.com/pack-insert/1815091a.pdf
(01.06.2010)
31

32

33
34

35

36

Online unter: http://tools.invitrogen.com/content/sfs/manuals/lipofectamine_man.pdf
(01.06.2010)
37

38
39

40