author's personal copy volume 117, no. 1, 15 september 2009
TRANSCRIPT

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Sm
Ka
b
c
a
ARRA
KATQDM
1
cfacpittipirprbti1
C
0d
Materials Chemistry and Physics 117 (2009) 148–155
Contents lists available at ScienceDirect
Materials Chemistry and Physics
journa l homepage: www.e lsev ier .com/ locate /matchemphys
ome benzotriazole derivatives as corrosion inhibitors for copper in acidicedium: Experimental and quantum chemical molecular dynamics approach
.F. Khaleda,∗, Sahar A. Fadl-Allahb, B. Hammouti c
Electrochemistry Research Laboratory, Chemistry Department, Faculty of Education, Ain Shams University, Roxy, Cairo, EgyptChemistry Department, Faculty of Science, Cairo University, Gamaa Street, 12613 Giza, EgyptLaboratoire de Chimie Appliquée & Environnement, Faculté des Sciences, Oujda, Morocco
r t i c l e i n f o
rticle history:eceived 17 February 2009eceived in revised form 3 May 2009ccepted 10 May 2009
a b s t r a c t
Three benzotriazole derivatives namely, 1-(Phenylsulfonyl)-1H-benzotriazole (PSB), 1-(3-Pyridinylsulfonyl)-1H-benzotriazole (3PSB) and 1-(2-Pyridinylsulfonyl)-1H-benzotriazole (2PSB)have been investigated for the corrosion of copper in 1 M HNO3 at different concentrations at 25 ± 1 ◦Cusing chemical (weight loss) and electrochemical (Tafel polarization method) measurements. Generally,
eywords:cid corrosion inhibitionafeluantum mechanicsFTolecular dynamics
inhibition efficiency of the investigated compounds was found to depend on the concentration and thenature of the inhibitors. Quantum chemical calculation results show that the benzotriazole ring andheteroatoms are the active sites of the three inhibitors. The adsorption behaviour of the studied inhibitorson copper surface has been studied using molecular dynamics (MD) method and density functionaltheory. The results indicated that the three benzotriazole derivatives could adsorb on the copper surfacefirmly through the benzotriazole ring and heteroatoms, the three inhibitors have excellent corrosion
inhibition performance.. Introduction
Copper is different from most of other metals in that it combinesorrosion resistance with high electrical and heat conductivity,ormability, machinability, and strength when alloyed, exceptt high temperatures. It is a relatively noble metal and does notorrode readily in acids, unless oxygen or other oxidizing agents areresent. Besides its nobility, the high corrosion resistance of copper
s also due to its ability to form passive oxide films or other protec-ive insoluble corrosion products [1,2]. The utilization of inhibitorso protect copper against corrosion is based on the ability of certainndividual chemical compounds, or mixtures, to reduce or to com-letely suppress the rate of corrosion processes, when introduced
n small concentrations into a corrosive medium. Inhibitors enableeduction of corrosion rate by influencing the kinetics of corrosionrocesses which are consistent with anodic and cathodic conjugate
eactions. A large number of organic compounds are known toe applicable as corrosion inhibitors for copper. These inhibitorshat used in the literature to inhibit copper corrosion in nitric acidnclude: imidazole [3], 1-phenyl-5-mercapto-1,2,3,4-tetrazole,,2,3,4-tetrazole (TTZ), 5-amino-1,2,3,4-tetrazole (AT) and∗ Corresponding author. Current address: Materials and Corrosion Laboratory,hemistry Department, Faculty of Science, Taif University, Taif, Saudi Arabia.
E-mail address: [email protected] (K.F. Khaled).
254-0584/$ – see front matter © 2009 Elsevier B.V. All rights reserved.oi:10.1016/j.matchemphys.2009.05.043
© 2009 Elsevier B.V. All rights reserved.
1-phenyl-1,2,3,4-tetrazole (PT) [4], bis[4-amino-5-hydroxy-1,2,4-triazol-3-yl]methane, bis[4-amino-5-hydroxy-1,2,4-triazol-3-yl]butane [5], N-(3-metoxyphenylaminomethyl) phthalimide, N-(3-methylphenylaminomethyl) phthalimide and N-(phenylami-nomethyl) phthalimide [6]. Most organic inhibitors contain at leastone polar group with an atom of nitrogen, sulphur and/or oxygen.These compounds possess an electron donor which can enhancechemical adsorption on a copper surface and will normally sup-press the cathodic reaction rate of a corrosion process. Inhibitionefficiency depends strongly on the structure of the moleculesunder a given condition. Benzotriazole (BTA) and its derivativeshave been studied extensively, and BTA has been proved to be ahighly efficient inhibitor for preventing the corrosion of copper andcopper-base alloys in neutral and alkaline media [7–9]. The pro-tective mechanisms were not completely understood. Generally,two types of mechanisms of inhibition were proposed. One wasthe formation of polymeric complexes with copper atoms (Cu+ andCu2+) depending on the applied conditions [8–11]. The other wasthe chemical adsorption of BTA on copper surfaces [7,12,13]. Somestudies suggested that the film formation and chemical adsorptionco-exist, but the dominant mechanism depends on solution pH andapplied potentials [14]. It was also reported that the presence of
BTA induced the formation of semiconductive copper oxides [15].This was possibly responsible for the improvement of corrosionresistance. Although BTA enables copper protection in variousaqueous environments, its efficiency decreases dramatically inacid solutions and high temperature conditions [7,12,16,17].
K.F. Khaled et al. / Materials Chemistry and Physics 117 (2009) 148–155 149
e of b
ermpattrto
2
ittewmc
wo
ut
spc
dt[iso
ft1Pamarat
Fig. 1. Chemical structur
In the absence of oxygen or other depolarizing agents copperxhibits very low rates of corrosion in non-oxidizing acids. The cor-osion of copper is higher in HCl than in H2SO4, particularly atoderate acid concentrations, because of the formation of com-
lexes such as CuCl2−. Oxidizing acids, such as nitric or chromiccid, attack copper rapidly because hydrogen is not a product ofhe overall reaction and the formation of solvated copper ions ishermodynamically favoured. In the presence of oxygen, the cor-osion of copper increases considerably in acidic solution. Underhese conditions, the overall reaction responsible for this rapid ratef copper corrosion is as follows:
Cu + 4H+ + O2 � 2Cu2+ + 2H2O
The rate of the anodic reaction is enhanced when complexingons such as NH3, are present in the solution, these species effec-ively reduce the concentration of uncomplexed copper ions andhus, the corrosion of copper takes place more rapidly [18]. How-ver, the rate of copper corrosion does not usually increase linearlyith the rate of the oxygen supply because the anodic reactionay be inhibited by the formation of a protective film during the
orrosion process [19].In order to support experimental data, theoretical calculations
ere conducted in order to provide molecular-level understandingf the observed experimental behaviour.
The major driving force of quantum chemical research is tonderstand and explain the functions of these benzotriazole deriva-ives in molecular terms.
Among quantum chemical methods for evaluation of corro-ion inhibitors, density function theory, DFT has shown significantromise [20] and appears to be adequate for pointing out thehanges in electronic structure responsible for inhibitory action.
In order to evaluate compounds as corrosion inhibitors andesign novel one, the relationship between the structural proper-ies of the molecules and their inhibition effects is being explored21,22]. Various organic compounds have been investigated for theirnhibiting behaviour and their molecular structures have been alsotudied by quantum chemical methods in order to elucidate therigins of their inhibition effect [23].
To be able to reveal new and effective corrosion inhibitorsor copper in 1 M HNO3 solutions, new benzotriazole deriva-ives namely, 1-(Phenylsulfonyl)-1H-benzotriazole (PSB),-(3-Pyridinylsulfonyl)-1H-benzotriazole (3PSB) and 1-(2-yridinylsulfonyl)-1H-benzotriazole (2PSB) have been useds possible inhibitors. The interactions between the inhibitor
olecules and the metal surfaces should by all means be explainednd understood in details. In examining these interactions, theo-etical approaches can be applied very usefully. Therefore, recentlyn increasing attention was seen on the involvement of theseheoretical approaches in corrosion studies [24].
enzotriazole derivatives.
The first objective of this work is to investigate the effect ofthe three benzotriazole derivatives on the corrosion of copperin 1 M HNO3 using chemical (weight loss) and electrochemical(Tafel extrapolation) methods. The second objective is to investi-gate the dependence of inhibition efficiency of these compounds ontheoretical chemical parameters such as the energies of the high-est occupied molecular orbital (EHOMO), the energy of the lowestunoccupied molecular orbital (ELUMO), the energy difference (�E)between EHOMO and ELUMO; net atomic charges, dipole moments(�) and total energies. The reactivity of these molecules was ana-lyzed through an evaluation of the Fukui indices. These parametershave been calculated using DFT (density function theory) methods.Also, explicit solvent simulations using molecular dynamics will beused to explore the adsorption mechanism of these benzotriazolederivatives on copper surface (1 1 0).
2. Experimental
The benzotriazole derivatives are obtained from Sigma–Aldrich Chemical Co.and their chemical structure are presented in Fig. 1. Copper specimens from John-son Mattey (Puratronic, 99.999%) were mounted in Teflon. An epoxy resin was usedto fill the space between Teflon and copper electrode. The circular cross-sectionalarea of the copper rod exposed to the corrosive medium that used in electrochem-ical measurements was (0.28 cm2). Weight loss measurements carried out usingcopper coupons of the same composition, each of size 2 cm length and 0.5 cm diam-eter (surface area = 6.5 cm2). Prior to all measurements, the coupons were etchedin a 7 M HNO3 solution for 15 s, then washed thoroughly with bidistilled water,degreased with acetone, washed again with bidistilled water, and finally dried atroom temperature.
The coupons were weighed and suspended in 100 ml of an aerated 1 M HNO3
solution containing the studied benzotriazole derivatives at the desired concen-trations for 36 h exposure period of time at 25 ± 1 ◦C. At the end of the tests, thecoupons were taken out, washed with bidistilled water, degreased with acetone,washed again with bidistilled water, dried, and then weighed using an analyticalbalance (precision: ±0.1 mg). Three measurements were performed in each caseand the mean value of the weight loss has been reported. The standard deviationof the observed weight loss was ±1%. The corrosion rate which is the weight lossper unit area, A per unit time, h (w, mg cm−2 h) as well as the inhibition efficiency(IEw%) over the exposure time period were calculated according to the followingequation:
IEw =(
1 − w
wo
)× 100 (1)
where wo and w are the weight loss without and with benzotriazole derivatives,respectively.
The electrochemical measurements were performed in a typical three-compartment glass cell consisted of the copper specimen as working electrode (WE),platinum counter electrode (CE), and a saturated calomel electrode (SCE) as the ref-erence electrode. The counter electrode was separated from the working electrodecompartment by fritted glass. The reference electrode was connected to a Luggincapillary to minimize IR drop. Solutions were prepared from bidistilled water of
resistivity 13 M� cm, the copper electrode was polished with different grit emerypapers up to 4/0 grade, cleaned with acetone, washed with bidistilled water andfinally dried.Electrochemical measurements were performed using Gamry Instrument Poten-tiostat/Galvanostat/ZRA. This includes a Gamry framework system based on theESA400, Gamry applications that include dc105 for dc corrosion measurements

1 istry and Physics 117 (2009) 148–155
af
p
3
adbsmDcttDtaa
eca
aimh
tc
f
Io
�
�
woo
fioo
f
f
f
weNctelbs
t
Table 1Corrosion rate in (mg cm−2 h−1), inhibition efficiency data obtained from weightloss measurements for copper in 1 M HNO3 solutions in the absence and presenceof various concentrations of benzotriazole derivatives at 25 ± 1 ◦C.
Concentration (M) Corrosion rate (mg cm−2 h−1) Ew(%)
Blank 0.341 –
PSB
10−4 0.200 41.355 × 10−4 0.072 78.8810−3 0.049 85.635 × 10−3 0.026 92.37
3PSB
10−4 0.208 39.005 × 10−4 0.094 72.4310−3 0.065 80.945 × 10−3 0.043 87.39
−4
50 K.F. Khaled et al. / Materials Chem
long with a computer for collecting data. Echem Analyst 4.0 software was usedor plotting, graphing and fitting data.
The Tafel current–potential curves were obtained by changing the electrodeotential automatically from −240 to +180 mVSCE with scan rate of 1 mV s−1.
. Computational details
There is no doubt that the recent progress in DFT has providedvery useful tool for understanding molecular properties and forescribing the behaviour of atoms in molecules. DFT methods haveecome very popular in the last decade due to their accuracy thatimilar to other methods in less time and with a smaller invest-ent from the computational point of view. In agreement with theFT, the energy of the fundamental state of polyelectronic systemsan be expressed through the total electronic density, and in facthe use of the electronic density instead of the wave function forhe calculation of the energy constitutes the fundamental base ofFT [25]. Atomic Fukui indices, which are obtained from the elec-
ron density, is useful in predicting which atoms in a moleculere most likely to suffer nucleophilic, electrophilic, or radialttacks.
The local reactivity of the molecules was analyzed through anvaluation of the Fukui indices [26]. These are measurments of thehemical reactivity, as well as an indicative of the reactive regionsnd the nucleophilic and electrophilic behaviour of the molecule.
The regions of a molecule where the Fukui function is largere chemically softer than the regions where the Fukui functions small, and by invoking the HSAB principle in a local sense, one
ay establish the behaviour of the different sites with respect toard or soft reagents.
The Fukui function f (�r) is defined as the derivative of the elec-ronic density �(�r) with respect to the number of electrons N at aonstant external potential �(�r):
(�r) =(
∂�(�r)∂N
)�(�r)
(2)
f the effects of relaxation associated with the addition or removalf electronic charges are not considered, then
+(�r) ≈ �LUMO(�r) (3)
−(�r) ≈ �HOMO(�r) (4)
here �LUMO(→r ) is the density of the lowest unoccupied molecular
rbital and �HOMO(�r) is density of the highest occupied molecularrbital [27].
The condensed Fukui functions [26] are found by taking thenite difference approximations from Mulliken population analysisf atoms in benzotriazole derivatives, depending on the directionf the electron transfer:
+k
= qk(N + 1) − qk(N) (for nucleophilic attack) (5)
−k
= qk(N) − qk(N − 1) (for electrophilic attack) (6)
ok = qk(N + 1) − qk(N − 1)
2(for radial attack) (7)
here qk is the gross charge of atom k in the molecule i.e. thelectron density at a point r in space around the molecule. The
corresponds to the number of electrons in the molecule. N + 1orresponds to an anion, with an electron added to the LUMO ofhe neutral molecule. N − 1 correspondingly is the cation with anlectron removed from the HOMO of the neutral molecule. All calcu-
ations are done at the ground-state geometry. These functions cane condensed to the nuclei by using an atomic charge partitioningcheme, such as Mulliken population analysis in Eqs. (5)–(7).An easy graphical display technique can also be used based onhe Fukui functions. Instead of calculating the molecular orbitals for
2PSB
10 0.219 35.775 × 10−4 0.102 70.0810−3 0.083 75.665 × 10−3 0.060 82.40
the neutral, cation, and anion, we can just add or subtract electronsfrom the molecular orbitals of the neutral molecule. This procedureis not as good as described above, but it does give a quick graphicaldisplay of the susceptibility of different kinds of attack. So ratherthan being a definitive calculation of a molecular property, freez-ing the molecular orbitals to those for the neutral molecule gives auseful graphical technique that can be rapidly applied.
The Fukui indices calculations were performed using MaterialsStudio DMol3 version 4.3.1 [28,29], a high quality quantum mechan-ics computer program (available from Accelrys, San Diego, CA).These calculations employed an ab initio, local density functional(LDF) method with a double numeric polarization (DNP) basis setand a Becke-Perdew (BP) functional. Dmol3 use a Mulliken popula-tion analysis [30].
The Discover molecular dynamics module in Materials Studio4.3 software from Accelrys Inc. [31] allows selecting a ther-modynamic ensemble and the associated parameters, definingsimulation time, temperature and pressuring and initiating adynamics calculation. The molecular dynamics simulation proce-dures have been described elsewhere [32].
The interaction energy ECu-inhibitor of the Cu surface with thebenzotriazole derivatives was calculated according to the followingequation:
ECu-inhibitor = Ecomplex − (ECu + Einhibitor) (8)
where Ecomplex is the total energy of the Cu crystal together with theadsorbed inhibitor molecule, ECu and Einhibitor are the total energyof the copper crystal and free inhibitor molecular, respectively. Thebinding energy of the inhibitor molecule is the negative value of theinteraction energy, Ebinding = −ECu-inhibition [33].
4. Results and discussions
4.1. Weight loss measurements
A series of weight loss measurements were carried out in 1 MHNO3 free solution and in solutions containing different concen-trations of benzotriazole derivatives. Corrosion rates of coppercoupons w in mg cm−2 h−1 and the inhibition efficiency (IEw) werecalculated using Eq. (1). The average corrosion rates, expressedin mg cm−2 h−1, are shown in Table 1. The results show that allbenzotriazole derivatives used inhibit the corrosion of copper in
1 M HNO3 solutions. The corrosion rate was found to depend onthe concentration of the inhibitor. Increasing the concentration ofeach benzotriazole derivatives increases the inhibition efficiencyIEw up to a maximum value (92.37% at 5 × 10−3 M for PSB, 87.39% at5 × 10−3 M for 3PSB and 82.40% at 5 × 10−3 M for 2PSB). This indi-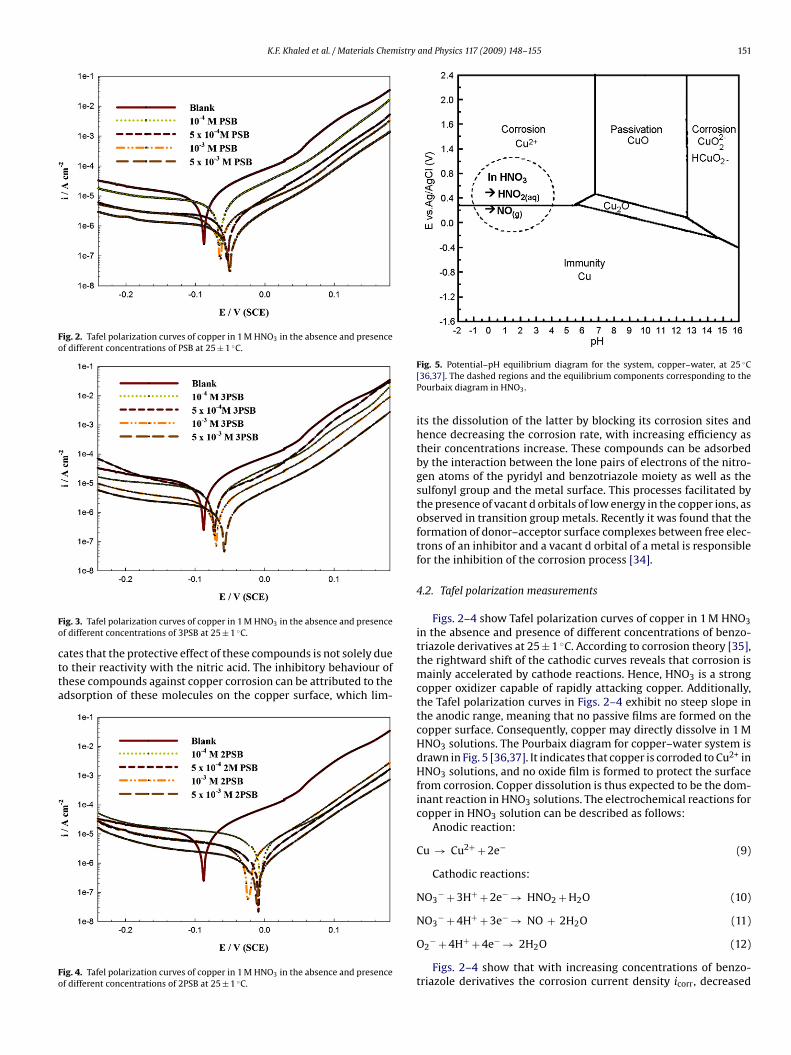
K.F. Khaled et al. / Materials Chemistry and Physics 117 (2009) 148–155 151
Fig. 2. Tafel polarization curves of copper in 1 M HNO3 in the absence and presenceof different concentrations of PSB at 25 ± 1 ◦C.
Fo
ctta
Fo
ig. 3. Tafel polarization curves of copper in 1 M HNO3 in the absence and presencef different concentrations of 3PSB at 25 ± 1 ◦C.
ates that the protective effect of these compounds is not solely dueo their reactivity with the nitric acid. The inhibitory behaviour ofhese compounds against copper corrosion can be attributed to thedsorption of these molecules on the copper surface, which lim-
ig. 4. Tafel polarization curves of copper in 1 M HNO3 in the absence and presencef different concentrations of 2PSB at 25 ± 1 ◦C.
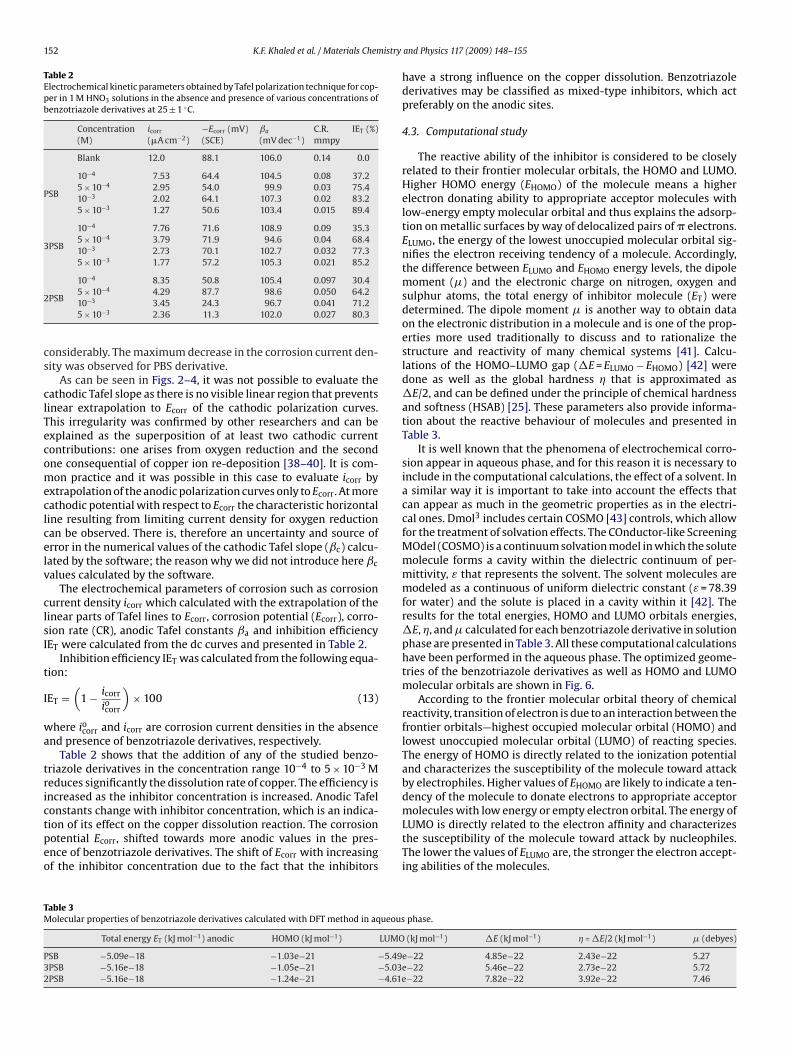
Fig. 5. Potential–pH equilibrium diagram for the system, copper–water, at 25 ◦C[36,37]. The dashed regions and the equilibrium components corresponding to thePourbaix diagram in HNO3.
its the dissolution of the latter by blocking its corrosion sites andhence decreasing the corrosion rate, with increasing efficiency astheir concentrations increase. These compounds can be adsorbedby the interaction between the lone pairs of electrons of the nitro-gen atoms of the pyridyl and benzotriazole moiety as well as thesulfonyl group and the metal surface. This processes facilitated bythe presence of vacant d orbitals of low energy in the copper ions, asobserved in transition group metals. Recently it was found that theformation of donor–acceptor surface complexes between free elec-trons of an inhibitor and a vacant d orbital of a metal is responsiblefor the inhibition of the corrosion process [34].
4.2. Tafel polarization measurements
Figs. 2–4 show Tafel polarization curves of copper in 1 M HNO3in the absence and presence of different concentrations of benzo-triazole derivatives at 25 ± 1 ◦C. According to corrosion theory [35],the rightward shift of the cathodic curves reveals that corrosion ismainly accelerated by cathode reactions. Hence, HNO3 is a strongcopper oxidizer capable of rapidly attacking copper. Additionally,the Tafel polarization curves in Figs. 2–4 exhibit no steep slope inthe anodic range, meaning that no passive films are formed on thecopper surface. Consequently, copper may directly dissolve in 1 MHNO3 solutions. The Pourbaix diagram for copper–water system isdrawn in Fig. 5 [36,37]. It indicates that copper is corroded to Cu2+ inHNO3 solutions, and no oxide film is formed to protect the surfacefrom corrosion. Copper dissolution is thus expected to be the dom-inant reaction in HNO3 solutions. The electrochemical reactions forcopper in HNO3 solution can be described as follows:
Anodic reaction:
Cu → Cu2+ + 2e− (9)
Cathodic reactions:
NO3− + 3H+ + 2e− → HNO2 + H2O (10)
NO3− + 4H+ + 3e− → NO + 2H2O (11)
O2− + 4H+ + 4e− → 2H2O (12)
Figs. 2–4 show that with increasing concentrations of benzo-triazole derivatives the corrosion current density icorr, decreased
152 K.F. Khaled et al. / Materials Chemistry
Table 2Electrochemical kinetic parameters obtained by Tafel polarization technique for cop-per in 1 M HNO3 solutions in the absence and presence of various concentrations ofbenzotriazole derivatives at 25 ± 1 ◦C.
Concentration(M)
icorr
(�A cm−2)−Ecorr (mV)(SCE)
ˇa
(mV dec−1)C.R.mmpy
IET (%)
Blank 12.0 88.1 106.0 0.14 0.0
PSB
10−4 7.53 64.4 104.5 0.08 37.25 × 10−4 2.95 54.0 99.9 0.03 75.410−3 2.02 64.1 107.3 0.02 83.25 × 10−3 1.27 50.6 103.4 0.015 89.4
3PSB
10−4 7.76 71.6 108.9 0.09 35.35 × 10−4 3.79 71.9 94.6 0.04 68.410−3 2.73 70.1 102.7 0.032 77.35 × 10−3 1.77 57.2 105.3 0.021 85.2
2
10−4 8.35 50.8 105.4 0.097 30.4
cs
clTecomeclcelv
clsI
t
I
wa
trictpeo
TM
P32
PSB5 × 10−4 4.29 87.7 98.6 0.050 64.210−3 3.45 24.3 96.7 0.041 71.25 × 10−3 2.36 11.3 102.0 0.027 80.3
onsiderably. The maximum decrease in the corrosion current den-ity was observed for PBS derivative.
As can be seen in Figs. 2–4, it was not possible to evaluate theathodic Tafel slope as there is no visible linear region that preventsinear extrapolation to Ecorr of the cathodic polarization curves.his irregularity was confirmed by other researchers and can bexplained as the superposition of at least two cathodic currentontributions: one arises from oxygen reduction and the secondne consequential of copper ion re-deposition [38–40]. It is com-on practice and it was possible in this case to evaluate icorr by
xtrapolation of the anodic polarization curves only to Ecorr. At moreathodic potential with respect to Ecorr the characteristic horizontaline resulting from limiting current density for oxygen reductionan be observed. There is, therefore an uncertainty and source ofrror in the numerical values of the cathodic Tafel slope (ˇc) calcu-ated by the software; the reason why we did not introduce here ˇc
alues calculated by the software.The electrochemical parameters of corrosion such as corrosion
urrent density icorr which calculated with the extrapolation of theinear parts of Tafel lines to Ecorr, corrosion potential (Ecorr), corro-ion rate (CR), anodic Tafel constants ˇa and inhibition efficiencyET were calculated from the dc curves and presented in Table 2.
Inhibition efficiency IET was calculated from the following equa-ion:
ET =(
1 − icorr
iocorr
)× 100 (13)
here iocorr and icorr are corrosion current densities in the absencend presence of benzotriazole derivatives, respectively.
Table 2 shows that the addition of any of the studied benzo-riazole derivatives in the concentration range 10−4 to 5 × 10−3 Meduces significantly the dissolution rate of copper. The efficiency isncreased as the inhibitor concentration is increased. Anodic Tafel
onstants change with inhibitor concentration, which is an indica-ion of its effect on the copper dissolution reaction. The corrosionotential Ecorr, shifted towards more anodic values in the pres-nce of benzotriazole derivatives. The shift of Ecorr with increasingf the inhibitor concentration due to the fact that the inhibitorsable 3olecular properties of benzotriazole derivatives calculated with DFT method in aqueous
Total energy ET (kJ mol−1) anodic HOMO (kJ mol−1) LUMO
SB −5.09e−18 −1.03e−21 −5.49PSB −5.16e−18 −1.05e−21 −5.03PSB −5.16e−18 −1.24e−21 −4.61
and Physics 117 (2009) 148–155
have a strong influence on the copper dissolution. Benzotriazolederivatives may be classified as mixed-type inhibitors, which actpreferably on the anodic sites.
4.3. Computational study
The reactive ability of the inhibitor is considered to be closelyrelated to their frontier molecular orbitals, the HOMO and LUMO.Higher HOMO energy (EHOMO) of the molecule means a higherelectron donating ability to appropriate acceptor molecules withlow-energy empty molecular orbital and thus explains the adsorp-tion on metallic surfaces by way of delocalized pairs of � electrons.ELUMO, the energy of the lowest unoccupied molecular orbital sig-nifies the electron receiving tendency of a molecule. Accordingly,the difference between ELUMO and EHOMO energy levels, the dipolemoment (�) and the electronic charge on nitrogen, oxygen andsulphur atoms, the total energy of inhibitor molecule (ET) weredetermined. The dipole moment � is another way to obtain dataon the electronic distribution in a molecule and is one of the prop-erties more used traditionally to discuss and to rationalize thestructure and reactivity of many chemical systems [41]. Calcu-lations of the HOMO–LUMO gap (�E = ELUMO − EHOMO) [42] weredone as well as the global hardness � that is approximated as�E/2, and can be defined under the principle of chemical hardnessand softness (HSAB) [25]. These parameters also provide informa-tion about the reactive behaviour of molecules and presented inTable 3.
It is well known that the phenomena of electrochemical corro-sion appear in aqueous phase, and for this reason it is necessary toinclude in the computational calculations, the effect of a solvent. Ina similar way it is important to take into account the effects thatcan appear as much in the geometric properties as in the electri-cal ones. Dmol3 includes certain COSMO [43] controls, which allowfor the treatment of solvation effects. The COnductor-like ScreeningMOdel (COSMO) is a continuum solvation model in which the solutemolecule forms a cavity within the dielectric continuum of per-mittivity, ε that represents the solvent. The solvent molecules aremodeled as a continuous of uniform dielectric constant (ε = 78.39for water) and the solute is placed in a cavity within it [42]. Theresults for the total energies, HOMO and LUMO orbitals energies,�E, �, and � calculated for each benzotriazole derivative in solutionphase are presented in Table 3. All these computational calculationshave been performed in the aqueous phase. The optimized geome-tries of the benzotriazole derivatives as well as HOMO and LUMOmolecular orbitals are shown in Fig. 6.
According to the frontier molecular orbital theory of chemicalreactivity, transition of electron is due to an interaction between thefrontier orbitals—highest occupied molecular orbital (HOMO) andlowest unoccupied molecular orbital (LUMO) of reacting species.The energy of HOMO is directly related to the ionization potentialand characterizes the susceptibility of the molecule toward attackby electrophiles. Higher values of EHOMO are likely to indicate a ten-dency of the molecule to donate electrons to appropriate acceptor
molecules with low energy or empty electron orbital. The energy ofLUMO is directly related to the electron affinity and characterizesthe susceptibility of the molecule toward attack by nucleophiles.The lower the values of ELUMO are, the stronger the electron accept-ing abilities of the molecules.phase.
(kJ mol−1) �E (kJ mol−1) � = �E/2 (kJ mol−1) � (debyes)
e−22 4.85e−22 2.43e−22 5.27e−22 5.46e−22 2.73e−22 5.72e−22 7.82e−22 3.92e−22 7.46
K.F. Khaled et al. / Materials Chemistry and Physics 117 (2009) 148–155 153
Fs
ditodtpiud
lo
lfdbTtb
Table 4Calculated Mulliken atomic charges and Fukui functions for benzotriazolederivatives.
Atom qN qN+1 qN−1 f +k
f −k
f ok
PSB
N (1) −0.209 −0.2060 −0.261 0.003 0.052 0.022N (2) −0.049 0.0250 −0.131 0.074 0.082 0.068N (3) −0.209 −0.1300 −0.296 0.079 0.087 0.078S (10) 0.492 0.5390 0.477 0.047 0.015 0.026O (11) −0.300 −0.243 −0.35 0.057 0.050 0.054O (12) −0.349 −0.299 −0.393 0.050 0.044 0.037
3PSB
N (1) −0.212 −0.216 −0.258 −0.004 0.046 0.011N (2) −0.048 −0.011 −0.084 0.037 0.036 0.036N (3) −0.207 −0.144 −0.271 0.063 0.064 0.064S (10) −0.499 −0.444 −0.506 0.055 0.007 0.031O (11) −0.294 −0.232 −0.339 0.062 0.045 0.053O (12) −0.348 −0.295 −0.380 0.053 0.032 0.043N (18) −0.202 −0.159 −0.210 0.043 0.008 0.070
2PSB
N (1) −0.224 −0.227 −0.258 −0.003 0.034 0.016N (2) −0.027 0.010 −0.058 0.037 0.031 0.059N (3) −0.205 −0.143 −0.257 0.062 0.052 0.082S (10) −0.518 −0.468 −0.525 0.050 0.007 0.028
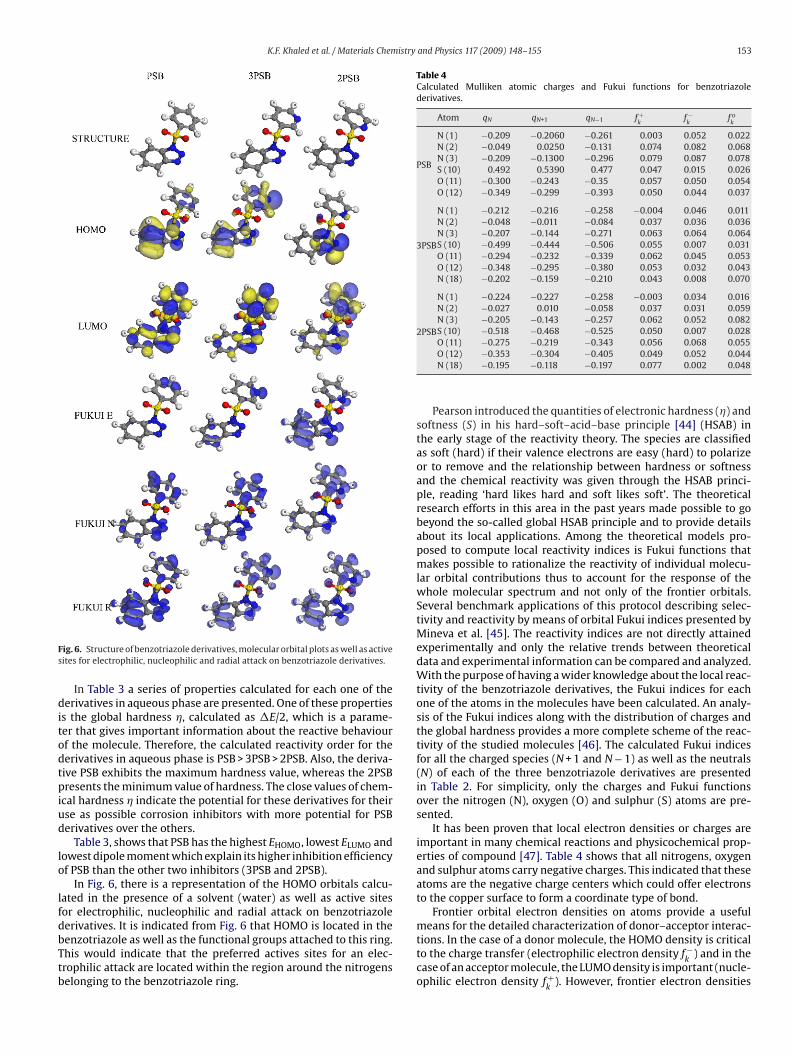
ig. 6. Structure of benzotriazole derivatives, molecular orbital plots as well as activeites for electrophilic, nucleophilic and radial attack on benzotriazole derivatives.
In Table 3 a series of properties calculated for each one of theerivatives in aqueous phase are presented. One of these properties
s the global hardness �, calculated as �E/2, which is a parame-er that gives important information about the reactive behaviourf the molecule. Therefore, the calculated reactivity order for theerivatives in aqueous phase is PSB > 3PSB > 2PSB. Also, the deriva-ive PSB exhibits the maximum hardness value, whereas the 2PSBresents the minimum value of hardness. The close values of chem-
cal hardness � indicate the potential for these derivatives for theirse as possible corrosion inhibitors with more potential for PSBerivatives over the others.
Table 3, shows that PSB has the highest EHOMO, lowest ELUMO andowest dipole moment which explain its higher inhibition efficiencyf PSB than the other two inhibitors (3PSB and 2PSB).
In Fig. 6, there is a representation of the HOMO orbitals calcu-ated in the presence of a solvent (water) as well as active sitesor electrophilic, nucleophilic and radial attack on benzotriazole
erivatives. It is indicated from Fig. 6 that HOMO is located in theenzotriazole as well as the functional groups attached to this ring.his would indicate that the preferred actives sites for an elec-rophilic attack are located within the region around the nitrogenselonging to the benzotriazole ring.O (11) −0.275 −0.219 −0.343 0.056 0.068 0.055O (12) −0.353 −0.304 −0.405 0.049 0.052 0.044N (18) −0.195 −0.118 −0.197 0.077 0.002 0.048
Pearson introduced the quantities of electronic hardness (�) andsoftness (S) in his hard–soft–acid–base principle [44] (HSAB) inthe early stage of the reactivity theory. The species are classifiedas soft (hard) if their valence electrons are easy (hard) to polarizeor to remove and the relationship between hardness or softnessand the chemical reactivity was given through the HSAB princi-ple, reading ‘hard likes hard and soft likes soft’. The theoreticalresearch efforts in this area in the past years made possible to gobeyond the so-called global HSAB principle and to provide detailsabout its local applications. Among the theoretical models pro-posed to compute local reactivity indices is Fukui functions thatmakes possible to rationalize the reactivity of individual molecu-lar orbital contributions thus to account for the response of thewhole molecular spectrum and not only of the frontier orbitals.Several benchmark applications of this protocol describing selec-tivity and reactivity by means of orbital Fukui indices presented byMineva et al. [45]. The reactivity indices are not directly attainedexperimentally and only the relative trends between theoreticaldata and experimental information can be compared and analyzed.With the purpose of having a wider knowledge about the local reac-tivity of the benzotriazole derivatives, the Fukui indices for eachone of the atoms in the molecules have been calculated. An analy-sis of the Fukui indices along with the distribution of charges andthe global hardness provides a more complete scheme of the reac-tivity of the studied molecules [46]. The calculated Fukui indicesfor all the charged species (N + 1 and N − 1) as well as the neutrals(N) of each of the three benzotriazole derivatives are presentedin Table 2. For simplicity, only the charges and Fukui functionsover the nitrogen (N), oxygen (O) and sulphur (S) atoms are pre-sented.
It has been proven that local electron densities or charges areimportant in many chemical reactions and physicochemical prop-erties of compound [47]. Table 4 shows that all nitrogens, oxygenand sulphur atoms carry negative charges. This indicated that theseatoms are the negative charge centers which could offer electronsto the copper surface to form a coordinate type of bond.
Frontier orbital electron densities on atoms provide a usefulmeans for the detailed characterization of donor–acceptor interac-
tions. In the case of a donor molecule, the HOMO density is criticalto the charge transfer (electrophilic electron density f −k) and in the
case of an acceptor molecule, the LUMO density is important (nucle-ophilic electron density f +
k). However, frontier electron densities

154 K.F. Khaled et al. / Materials Chemistry
Table 5Interaction and binding energy of the benzotriazole derivatives on copper (1 1 0)surface.
Inhibitor ECu-inhibitor (kJ mol−1) Ebinding (kJ mol−1)
P32
ci
mofsptapPNp
a3adbAi
SB −328.5 328.5PSB −298.6 298.6PSB −278.3 278.3
an strictly be used only to describe the reactivity of different atomsn the same molecule.
For a finite system such as an inhibitor molecule, when theolecule is accepting electrons, one has f +
k, the index for nucle-
philic attack; when the molecule is donating electrons, one has−k
, the index for electrophilic attack. An analysis of Fukui indiceshown in Table 4, in which only the largest values are presented, it isossible to be observed that in each one of the benzotriazole deriva-ives, the nitrogen atoms in the benzotriazole ring (N1, N2 and N3)re the most susceptible sites for electrophilic attacks. These sitesresent the highest values of f −
kwhich range from 0.052 to 0.087. In
SB the phenyl ring offers a susceptible site for electrophilic attack.evertheless, in case of both 3PSB and 2PSB the presence of theyridyl ring enhances the nucleophilic attack character f +
k.
The molecular dynamics simulations are performed to study thedsorption behaviour of the studied benzotriazole derivatives (PSB,PSB and 2PSB) on copper (1 1 0) surface. The values of the inter-ction energy and the binding energy of the three benzotriazole
erivatives on copper (1 1 0) surface are listed in Table 5. As cane seen from Table 5 that the binding energy has a positive value.s the value of the binding energy increases, the more easily thenhibitor adsorbs on the metal surface, the higher the inhibition
Fig. 7. Mode of adsorption of the benzotriazole derivative on copper (1 1 0).
and Physics 117 (2009) 148–155
efficiency [33]. PSB has the highest binding energy comparing to3PBS and 2PSB to the copper surface that found during the sim-ulation process. High values of binding energy obtained with PSBmolecules explain its highest inhibition efficiency from the theo-retical point of view. Benzotriazole derivatives studied in this workhave a number of lone pairs of electrons on atoms like S, N and Oas well as � electron clouds on the aromatic rings. This will makeit possible to provide electrons to the unoccupied orbitals of cop-per oxide surface in HNO3 solutions that form a stable coordinationbonds. Therefore the studied benzotriazoles are likely to adsorb onthe copper surface and protect it from corrosion.
Fig. 7 shows the equilibrium configuration of the benzotriazolederivatives. According to the equilibrium configuration of the threebenzotriazole derivatives adsorbed on copper (1 1 0) surface, wecan draw a conclusion that the studied benzotriazole derivativescan be adsorbed on the copper surface through the benzotria-zole ring and heteroatoms. In this way, the exposed part of coppersurface can be reduced by the covering of the benzotriazole ringand heteroatoms, consequently preventing the surface from acidcorrosion.
4.4. Inhibition mechanism of benzotriazole derivatives
The transition of metal/solution interface from a state of activedissolution to the passive state is attributed to the adsorption ofthe inhibitor molecules and the metal surface, forming a protectivefilm. The rate of adsorption is usually rapid and hence, the reactivemetal surface is shielded from the aggressive environment [48].Adsorption of benzotriazole derivatives can be described by twomain types of interactions: physical adsorption and chemisorption.In general, physical adsorption requires the presence of both theelectrically charged surface of the metal and charged species insolution. The surface charge of the metal is due to the electric fieldexisting at the metal/solution interface. A chemisorption process,on the other hand, involves charge sharing or charge transfer fromthe inhibitor molecules to the metal surface to form a coordinatetype of a bond. This is possible in the case of a positive as well as anegative charge of the surface. The presence of a transition metal,having vacant, low-energy electron orbitals (Cu+ and Cu2+ in ourcase) and of an inhibitor with molecules having relatively looselybound electrons or heteroatoms with a lone pair of electrons is nec-essary [15]. Generally, two types of mechanisms of inhibition wereproposed. One was the formation of polymeric complexes with cop-per ions (Cu+ and Cu2+) depending on the applied conditions [8,9].The other was the chemical adsorption of benzotriazoles on coppersurfaces [7,13]. Benzotriazole derivatives in this study present simi-lar inhibition efficiencies. PSB gives the highest inhibition efficiencyamong 3PSB and 2PSB. The high inhibition efficiency of PSB is dueto the presence of the phenyl ring which has sites susceptible toelectrophilic attack as well as low HOMO energy, Low �E and lowhardness. The other was the chemical adsorption of benzotriazoleson copper surfaces [7,13].
The structure of copper–benzotriazole complexes that thoughtto be formed on copper surface in acid medium, according to thestudies of the structure of the Cu(I) [49] and Cu(II) [50] azole com-plexes, it is concluded that the bonding occurs through N atomsand in the presence of other heteroatoms like sulphur they can alsoparticipate in bonding [51,52], and that the coordination numbercan be as high as four. It is found that a stable Cu2O is formedonly in acidic solution of pH over 2. Nevertheless, since the pro-ton H+ is continuously consumed to the hydrogen gas H2 during
the Cu dissolution, the pH value near the Cu electrode in molarnitric acid instantaneously jump from 1 to greater values. Thusit is acceptable to think that Cu2O can be metastably formed onthe Cu surface even at open circuit potential in molar nitric acid[3]. Adsorption in this case is assisted by hydrogen bond formation
istry
bobTNtiTtsrr
5
2
3
4
5
R
[
[
[[[[
[[
[[[[
[[
[[
[[[[[[[[
[
[[[[[
[
[[[
[
[[48] C.Y. Chao, L.F. Lin, D.D. Macdonald, J. Electrochem. Soc. 128 (1981) 1187.
K.F. Khaled et al. / Materials Chem
etween benzotriazole derivatives and the intermediates. The typef intermediates that formed on Cu surface in molar nitric acid cane explained according to pourbaix diagram presented in Fig. 5.his type of adsorption should be more prevalent for protonated-atoms, because the positive charge on the N-atom is conduc-
ive to the formation of hydrogen bonds. Unprotonated N-atomsn benzotriazole derivatives may adsorb by direct chemisorption.here is a noticeable discrepancy regarding the surface interac-ions, whether pure copper or copper oxides participate. It is alsotill not completely clear what is the most determining parameteresponsible for efficient inhibition thus leaving space for furtheresearch.
. Conclusion
The following results can be drawn from this study:
1. Experimental investigations of the studied benzotriazole deriva-tives show that they reduce the corrosion rate of copperremarkably as their concentration increase.
. Polarization studies show that this class of compounds acts asmixed-type inhibitors with predominant anodic effect.
. The presence of nitrogen and other heteroatoms such as oxy-gen and sulphur, those are able to bond with copper, as well asthe presence of � electrons enables the interaction and bondingbetween benzotriazole derivatives and copper surface.
. The quantum mechanical approach may well be able to foretellmolecule structures that are better for corrosion inhibition.
. Molecular modeling techniques incorporating molecularmechanics and molecular dynamics can be used to simulatethe adsorption from 1 M HNO3 solution of some benzotriazolederivatives on copper (1 1 0) surface.
eferences
[1] S.B. Adeloju, H.C. Hughes, Corros. Sci. 26 (1986) 851.[2] T. Suter, E.M. Moser, H. Bohni, Corros. Sci. 34 (1993) 1111.[3] W.J. Lee, Mater. Sci. Eng. A 348 (2003) 217.[4] M. Mihit, R. Salghi, S. El Issami, L. Bazzi, B. Hammouti, El. Ait Addi, S. Kertit,
Pigment Resin Tech. 35 (2006) 151.[5] M.M. El-Naggar, Corros. Sci. 42 (2000) 773.
[6] A.S. Fouda, A. Abd El-Aal, A.B. Kandil, Desalination 201 (2006) 216.[7] M.M. Musiani, G. Mengoli, J. Electroanal. Chem. 217 (1987) 187.[8] S.F.L.A. Da Costa, S.M.L. Agostinho, J.C. Rubim, J. Electroanal. Chem. 295 (1990)203.[9] V. Brusic, M.A. Frisch, B.N. Eldridge, F.P. Novak, F.B. Kauman, B.M. Rush, G.S.
Frankel, J. Electrochem. Soc. 138 (1991) 2253.
[
[[[
and Physics 117 (2009) 148–155 155
10] G. Xue, J. Ding, Appl. Surf. Sci. 40 (1990) 327.[11] H.C. Shih, R.J. Tzou, Corros. Sci. 35 (1993) 479.12] G.A. Hope, C.A. Davis, D.P. Schweinsberg, Proceeding of Ninth Australasian Elec-
trochemistry Conference, Part I, 07-1, University of Wollongong, Wollongong,NSW, Australia, 1994.
13] G. Lewis, Corrosion 34 (1978) 424.14] R. Youda, H. Nishihara, K. Aramaki, Electrochim. Acta 35 (1990) 1011.15] A. Aruchamy, A. Fujishima, J. Electroanal. Chem. 266 (1989) 397.16] Y.C. Wu, P. Zhang, H.W. Pickering, D.L. Ahara, J. Electrochem. Soc. 140 (1993)
2489–2791.[17] J. Penninger, K. Woppermann, J.W. Schultze, Werkst. Korros. 38 (1987) 649.18] H. Leidheiser, The Corrosion of Copper Tin, and Their Alloys, Wiley, N.Y., 1971.19] S. Sathiyanarayanan, S.P. Manoharan, G. Rajagopal, K. Balakrishnan, Br. Corros.
J. 27 (1992) 72.20] N. Lopez, F. Illas, J. Phys. Chem. B 102 (1998) 1430.21] A.G. Gad Allah, H. Moustafa, J. Appl. Electrochem. 22 (1992) 644.22] F. Bentiss, M. Traisnel, H. Vezin, M. Lagrenee, Corros. Sci. 45 (2003) 371.23] W. Durnie, R. De Marco, B. Kinsella, A. Jefferson, B. Pejcic, J. Electrochem. Soc.
152 (2005) B1.24] K.F. Khaled, Electrochim. Acta 54 (2009) 4345.25] J. Andrés, J. Beltran, Química Teórica y Computacional, Universitat Jaume I,
Castellón de la Plana, Espana, 2000.26] W. Yang, W.J. Mortier, J. Am. Chem. Soc. 108 (1986) 5708.27] F. Méndez, M. Galván, A. Garritz, A. Vela, J. Gázquez, J. Mol. Struct. 277 (1992)
1981.28] B. Delley, J. Chem. Phys. 92 (1990) 508.29] B. Delley, J. Chem. Phys. 113 (2000) 7756.30] R.S. Mulliken, J. Chem. Phys. 23 (1995) 1833.31] J. Barriga, B. Coto, B. Fernandez, Tribol. Int. 40 (2007) 960.32] K.F. Khaled, J. Solid State Electrochem., (2009) doi:10.1007/s10008-009-0845-y.33] S. Xia, M. Qui, L. Yu, F. Lui, Corros. Sci. 50 (2008) 2021.34] D. Zhang, Q. Cai, X. He, L. Gao, G. Soon Kim, Mater. Chem. Phys. 114 (2009) 612.35] D.A. Jones, Principles and Prevention of Corrosion, second ed., Prentice Hall,
Upper Saddle River, NJ, 1983.36] M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, NACE,
Houston, TX, 1975.37] H.E. Johnson, J. Leja, J. Electrochem. Soc. 112 (1965) 638.38] G. Quartarone, T. Bellomi, A. Zingales, Corros. Sci. 45 (2003) 722.39] W. Schltze, K. Wippermann, Electrochim. Acta 32 (1987) 823.40] T.N. Anderson, M.H. Ghandehari, H. Eryng, J. Electrochem. Soc. 128 (1975) 1580.41] V.S. Sastri, Corrosion Inhibitors—Principles and Applications, Wiley, Chichester,
England, 1998.42] J.B. Foresman, AE. Frisch, Exploring Chemistry with Electronic Structure Meth-
ods, 2nd ed., Gaussian, Inc., Pittsburgh, PA, 1996.43] A. Klamt, G. Schüürmann, J. Chem. Soc., Perkin Trans. 2 (1993) 799.44] R.G. Pearson, J. Am. Chem. Soc. 85 (1963) 3533.45] T. Mineva, V. Parvanov, I. Petrov, N. Neshev, N. Russo, J. Phys. Chem. A 105 (2001)
1959.46] J. Cruz, L.M. Martínez-Aguilera, R. Salcedo, M. Castro, Int. J. Quant. Chem. 85
(2001) 546.47] M. Karelson, V.S. Lobanov, Chem. Rev. 96 (1996) 1027.
49] J.-P. Zhang, Y.-Y. Lin, X.-C. Huang, X.-M. Chen, Copper(I), J. Am. Chem. Soc. 127(2005) 5495.
50] F.A. Cotton, C.A. Murillo, X. Wang, Inorg. Chem. Commun. 1 (1998) 281.51] M. Fonsati, F. Zucchi, G. Trabanelli, Electrochim. Acta 44 (1998) 311.52] L. Larabi, O. Benali, S.M. Mekelleche, Y. Harek, Appl. Surf. Sci. 253 (2006) 1371.