attempts to synthesize vinyl thiazoles by geraldine...
TRANSCRIPT

Attempts to synthesize vinyl thiazoles
Item Type text; Thesis-Reproduction (electronic)
Authors Ciko, Geraldine Ann, 1941-
Publisher The University of Arizona.
Rights Copyright © is held by the author. Digital access to this materialis made possible by the University Libraries, University of Arizona.Further transmission, reproduction or presentation (such aspublic display or performance) of protected items is prohibitedexcept with permission of the author.
Download date 16/07/2018 00:07:41
Link to Item http://hdl.handle.net/10150/318492

ATTEM PTS TO SYNTHESIZE VINYL THIAZOLES
by
G erald ine Ann Ciko
A T hesis Subm itted to the F acu lty of the
DEPARTM ENT OF CHEMISTRY
In P a r t ia l F u lfillm en t of the R equ irem en ts F o r the D egree of
MASTER OF SCIENCE
In the G raduate College
THE UNIVERSITY O F ARIZONA
19 66

STATEMENT BY AUTHOR
T his th e s is has been subm itted in p a rtia l fu lfillm ent of req u ire m e n ts fo r an advanced degree at The U n iversity of A rizona and is deposited in the U n iversity L ib ra ry to be m ade availab le to b o rro w e rs under ru le s of the L ib ra ry .
B rie f quotations from th is th e s is a re allow able without spec ia l p e rm iss io n , provided that accu ra te acknowledgm ent of sou rce is m ade. R equests fo r p e rm iss io n for extended quotation from o r rep roduction of th is m an u scrip t in whole o r in p a rt m ay be g ran ted by the head of the m a jo r departm en t o r the Dean of the G raduate C ollege when in his judgm ent the p roposed u se of the m a te r ia l is in the in te re s ts of sch o larsh ip . In a ll o ther in stan ces , how ever, p e rm iss io n m ust be obtained from the author.
SIGNED
APPROVAL BY THESIS DIRECTOR
This th e s is has been approved on the date shown below:
A ssocia te P ro fe s s o r of C h em istryJAMES E. MULVANEY
s t i

ACKNOWLEDGMENT
I would like to thank D r. Jam es M ulvaney fo r d irec tin g th is
w ork and for the in te re s t and patience he has shown throughout my
two y e a rs of g raduate study.
I would also like to thank m y p a re n ts , w ithout whose love,
devotion, and encouragem ent m y attainm ent of th is degree would
not have been p oss ib le .

TABLE OF CONTENTS
Page
LIST OF TABLES . vi
ABSTRACT * * * • * * ♦ * ♦ ♦ * ♦ ♦ • * ♦ * ♦ * « ♦ « » ♦ • » ♦ » ♦ ♦ ♦ • ♦ * • # » * y ii
INTRODUCTION 1
DISCUSSION OF RESULTS , . , , ................................... 7
EXPERIM ENTAL 19
F r ie d e l-C ra f ts R eaction of C hloroacety l C hlorideand E thylene 19
M ethyl Vinyl Ketone and T hioacetam ide with SulfurylC hloride ............... 20
M ethyl Vinyl Ketone and T hioacetam ide with Iodine . . . . . 21 M ethyl Vinyl Ketone and T h ioacetam ide with Thionyl
C h lo rid e ...................................................... 21A cry lo n itr ile and Hydrogen Sulfide ........................ 21A cry lam ide and P hosphorus P en tasu lfide in Xylene . . . . . 22 A cry lam ide and Phosphorus P en tasu lfide in C hloroform . . 23 M ethacrylam ide and Phosphorus P en tasu lfide in
C hloroform ....................................... 24T hioacry lam ide and C hloroacetaldehyde . .................. 24T hioacry lam ide and C h lo roacetal in E thanol . . . . . . . . . . 25A cry lam ide , P hosphorus P en tasu lfide , and
C h lo ro a c e to n e ................................................ . . . . . . . . 25T h ioacry lam ide and C h lo roaceta l in H ydrochloric
A cid ...................... 26T hioacry lam ide and C h lo roacetal w ith A nhydrous
O xalic Acid ........................... 26T hioacry lam ide and (phloroacetal in E thanol
(Acid Catalyzed) . . . . . . . . . . . . . . . . . . . . . . . . . 27T hiom ethacry lam ide and C h lo roacetal with A nhydrous
O xalic Acid . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27T hiom ethacry lam ide and C h lo roaceta l in
D im ethy lfo rm am ide , ...................28T h ioacry lam ide and C yclopentadiene ........................ . 28T hiom ethacry lam ide and Cyclopentadiene . . . . . . . . . . . 28
iv

V
TABLE OF CONTENTS - - (Continued)
Page
T hiom ethacry 1-amide with H ydrochloric Acid inE t h a n o l ............................................. 29
B ase-C ata ly zed P o ly m eriza tio n of T h io a c ry la m id e ................. 29B ase -C ata ly zed P o ly m eriza tio n of T h iom ethacry lam ide . . . 30
REFERENCES .......................................................................................31

LIST OF TABLES
Table Page
1. Sum m ary of A ttem pts to Synthesize Vinyl and2-Isopropeny l T h iazo les . . . . . . . . . . . . . . . . . . . . . . 11
v i

ABSTRACT
Reaction of acry lam ide with phosphorus pentasu lfide in ch lo ro
form y ields th ioacry lam ide (I, 45. 9%). U nder the sam e conditions
m ethacry lam ide y ields th iom ethacry lam ide (II, 44%). When reac ted
with chloroacetaldehyde o r ch loroacetaldehyde diethyl ace ta l, both
th ioam ides failed to yield th iazo les .
P o ly m eriza tio n of (II) with po tassium t-bu tox ide yielded p r i
m a rily sim ple vinyl po lym er. P o ly m eriza tio n of (I) under the sam e
conditions yielded a po lym er of undeterm ined s tru c tu re .
CH =NH
I II

INTRODUCTION
Until recen tly , the m ost w idely studied vinyl m onom ers and
po lym ers w ere those of s ty ren e and its d e riv a tiv es . T hese studies
have yielded m uch in form ation concerning the effect of substituen ts on
the double bond on m onom er rea c tiv ity . The m ost d ire c t m eans of
de term in ing m onom er rea c tiv ity is through copolym erization e x p e ri
m en ts, w hereby the tendency of the active chain end, w hether it be
rad ic a l, carbonium ion, o r carbanion , to add to its own m onom er o r
the second m onom er can be m easu red .
A ssum ing the following reac tio n s to take p lace during a
copolym erization
-M • + M 11
+ M2
-M.
-Mg' + M 2T-m 2-
-M -
-m 2-+ m 2 -M2-
and defining M and M9 as m onom er 1 and m onom er 2, resp ec tiv e ly ,
ku k22r i aS ” k------' and Tg as — ----- , the copolym er com position equation
12 21
dMt M l r 1[M1] + [M2]"
dM2 M2 r 2 [M2 J + Im J
1
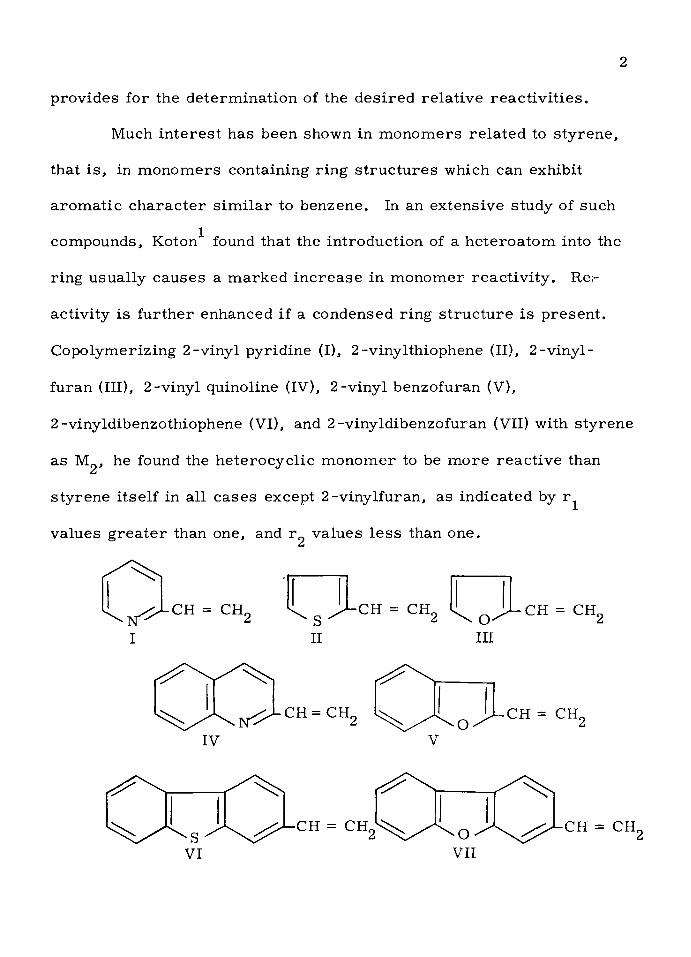
provides fo r the de te rm ina tion of the d e s ire d re la tiv e re a c tiv itie s .
Much in te re s t has been shown in m onom ers re la te d to s ty ren e ,
that is , in m onom ers containing ring s tru c tu re s which can exhibit
a ro m a tic c h a ra c te r s im ila r to benzene. In an ex tensive study of such
com pounds, Koton* found that the in troduction of a h e te ro a to m into the
ring usually causes a m arked in c re a se in m onom er re a c tiv ity . R e
activ ity is fu rth e r enhanced if a condensed rin g s tru c tu re is p resen t.
Co po lym eriz ing 2 -vinyl pyrid ine (I), 2 -vinylthiophene (II), 2 -v iny l-
fu ran (III), 2 -vinyl quinoline (IV), 2 -vinyl benzofuran (V),
2 -vinyldibenzothiophene (VI), and 2 -v inyld ibenzofuran (VII) with s ty ren e
as Mg, he found the h e te ro cy c lic m onom er to be m ore rea c tiv e than
s ty ren e its e lf in a ll cases except 2 -v iny lfuran , as ind icated by r^
values g re a te r than one, and r^ values le s s than one.
I
CH = CH,SII
-CH = CHrO '
III
CH = CHr
IV
CH = CHr CH = CH
CH = CH CH = CH,

In addition, Koton found th a t m onom ers containing condensed ring
s tru c tu re s w ere m ore rea c tiv e than those containing only one ring .
2K oton 's re s u lts su b stan tia te those rep o rte d e a r l ie r by W alling on
vinyl pyrid ine and v inyl thiophene.
While the tren d tow ard in c re a se d re a c tiv ity of h e te ro cy c lic
m onom ers is not confined to copo lym erization with s ty re n e , the use
of o ther m onom ers as y ie lds re s u l ts not quite as consisten t.
3 4F ran k ’ showed that while 2 -, 3 -, and 4 -vinyl pyrid ine and se v e ra l
a lky lated d e riv a tiv es of th ese copolym erize fa s te r with butadiene than
does s ty ren e , 2 -v inylfuran , and 2 -vinylthiophene copolym erize slow er5
than s ty ren e with the sam e com onom er, and K am enar found.that the
copo lym erization of 2 -v iny lfu ran with vinylidene ch lo ride p roceeded at
a slow er ra te than did the ho mo po lym eriza tion of e ith e r m onom er. In
the case of the 2 -v inylth iophene-bu tad iene sy stem , how ever, Meehan^
showed that, while the copo lym erization p roceeded at a slow er ra te
than did the s ty ren e -butadiene system ,. 2 - v inyl thiophene e n te rs the
chain at a fa s te r ra te than , does s ty ren e , since butadiene - s ty ren e
(75:25) copolym ers a t 70-80% conversion have s ty ren e constan ts of
20 .3 -21 .2% , while 2 -vinyl thiophene e n te rs the copolym er a t a ra te
approxim .ately p ro p o rtio n a l to i ts concentration, in the m onom er m ix
tu re (25%).
The fu ran rin g apparen tly deac tiva tes the vinyl group in7
copo lym erization re a c tio n s . B orrow s stud ied the effect of fu ran

analogs of e innam onitrile on the po lym eriza tion of s ty re n e , and found
that th ese compounds inhib it the po lym eriza tion in the p resen c e of a ir
and re ta rd in the absence of a i r . Koton* also found th a t the p ro p e rtie s
of vinyl fu ran hom opolym ers a re dependent on the ava ilab ility of
m o lecu la r oxygen. In the p resen c e of oxygen, a soft, low m elting
po lym er was obtained, while in the absence of oxygen, f re e ra d ic a l
in itia tion .y ie lded a h a rd , infusib le po lym er., Koton p roposed that, in
th is case , the double bonds in the fu ran also po lym erize , form ing a
th ree -d im en s io n a l, highly c ro ss - lin k e d po lym er.
The addition of a second o r th ird h e te ro a to m fu r th e r enhances
activ ity , as has been shown by the copo lym erization of s ty re n e with
2, 4 -d im e th y l- 6 -v in y l-s - tr ia z in e and 2 - dim ethyl am ino- 4 -vinylpy r im i -
8dine .
A nother a re a of in te re s t in the study of vinyl he te rocyc les, is
th a t of b io log ical ac tiv ity . A lthough th e re a re a g re a t m any n a tu ra lly -
o ccu rrin g po lym ers which exhibit b io log ical activ ity , notably the
p ro te in s and nucleic ac id s, th ese a re c la ss if ied as condensation poly
m e rs , and vinyl po lym ers exhibiting s im ila r activ ity a re r a r e , if
indeed, ex isten t a t a ll.
Some syn thetic vinyl p o lym ers, how ever, do show bio logical
ac tiv ity . Polyvinyl py rro lidone (VIII) has been used in the p rep a ra tio ng
. of syn thetic blood p lasm a , although the rea so n s fo r i ts e ffectiveness
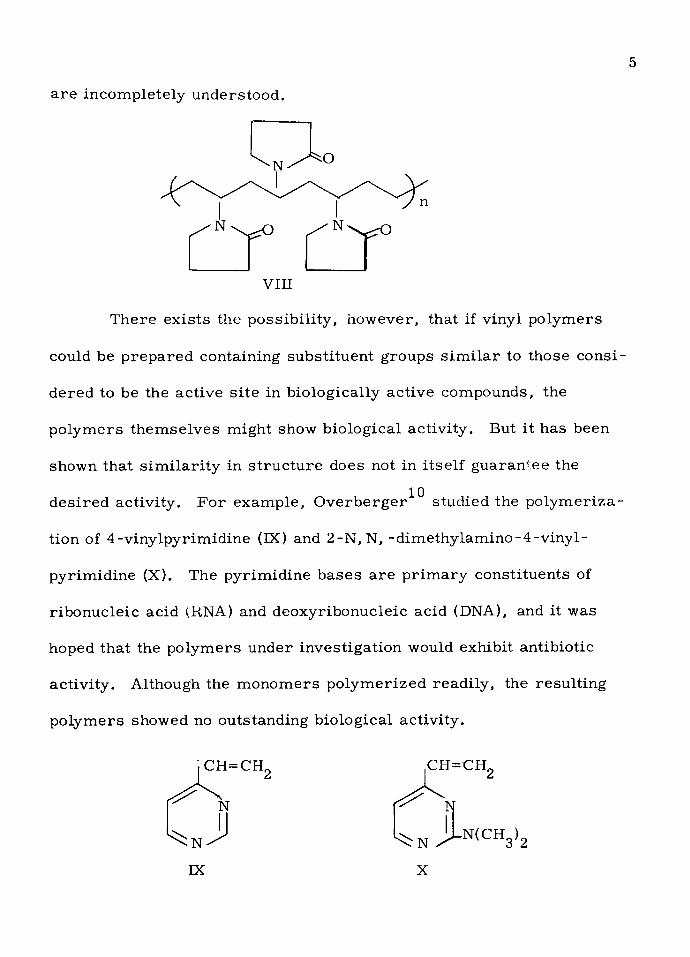
5
a re incom pletely understood .
VIII
T here ex is ts the p o ssib ility , how ever, that if vinyl po lym ers
could be p rep a red containing substituen t groups s im ila r to those co n si
dered to be the active s ite in b io logically active com pounds, the
po lym ers them se lves m ight show b io log ical ac tiv ity . But it has been
shown that s im ila r ity in s tru c tu re does not in i ts e lf guaran tee the
d e s ired activ ity . F o r exam ple, Overberger"*- ̂ studied the p o ly m eriza
tion of 4 -v inylpyrim id ine (IX) and 2-N , N, - d im ethy lam ino-4 - v iny l-
pyrim id ine (X). The pyrim id ine b ases a re p rim a ry constituen ts of
ribonucleic acid (RNA) and deoxyribonucleic acid (DNA), and it was
hoped tha t the po lym ers under investigation would exhibit antib iotic
ac tiv ity . Although the m onom ers po lym erized read ily , the resu ltin g
po lym ers showed no outstanding b io logical activ ity .
CH—CH,
X
i CH=CH,
rx
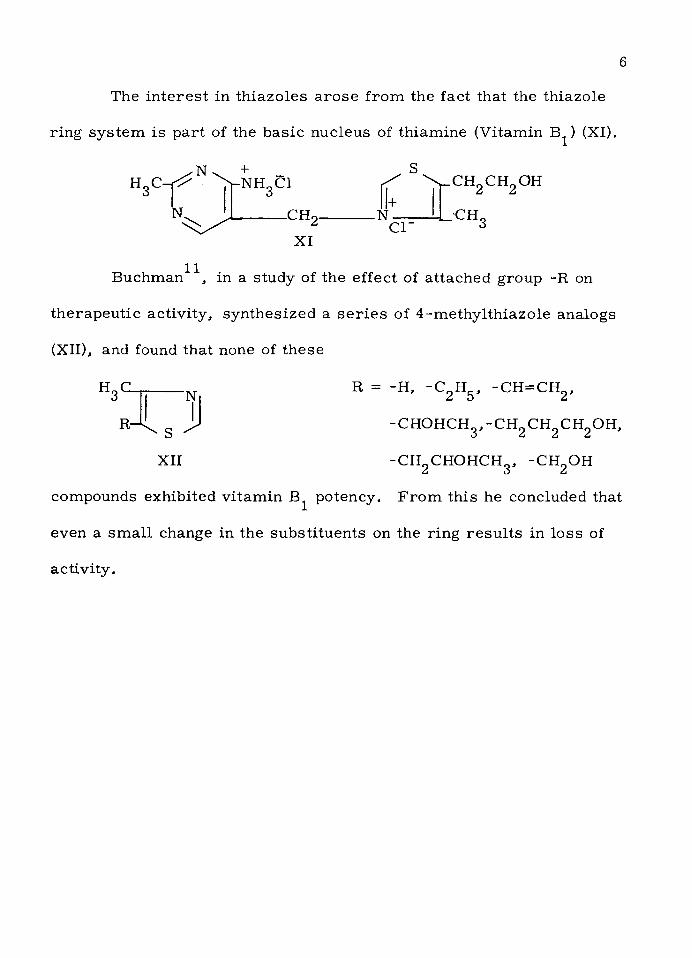
The in te re s t in th iazo les a ro se from the fact that the th iazole
ring sy stem is p a rt of the basic nucleus of th iam ine (V itam in B^) (XI).
H3C"TNK + _
y-NHgCl
N. -CH2
XI
+.N
CT
.CH2 CH2 OH
- CH3
11Buchman , in a study of the effect of a ttached group -R on
th e rap eu tic activ ity , syn thesized a s e r ie s of 4 -m ethy lth iazo le analogs
(XII), and found that none of these
H3C-
R-
‘N
- S -
XII
R = -H, -C 2 H5 , -C H =C H 2 ,
- c h o h c h 3 , - c h 2 c h 2c h 2o h ,
-C H 2 CHOHCH3, -CHgOH
compounds exhibited v itam in B^ potency. F ro m th is he concluded that
even a sm a ll change in the sub stitu en ts on the ring re s u lts in lo ss of
activ ity .

DISCUSSION
Although the syn thesis of vinyl th iazo les and 2 -isopropeny l
th iazo les is rep o rte d in the l i te ra tu re , * * * ̂ these m ethods involve as
the final step the dehydration of a lcohols, the dehydrohalogenation of
halogenated d e riv a tiv e s , o r the p y ro ly sis of e s te r s . This work deals
with a ttem pts to p re p a re vinyl th iazo les and 2 -isop ropeny l th iazo le
from compounds a lread y containing the v inylic bond, and, finally , the
po lym eriza tion of these com pounds.
p ro p er th ioam ides and carbonyl com pounds, it is possib le to obtain
an un lim ited num ber of substitu ted o r unsubstitu ted th iazo le s .
The m ost gen era lly applicable m ethod of th iazo le syn thesis is
14the reac tio n of th ioam ides with u-halo carbonyl com pounds. The
m echan ism appears to be as follows:
w here -R , -R , and -R m ay be H, alkyl, a ry l, e tc . By the use of the
7

T hioform am ide is rep o rte d in the l i t e r a t u r e ^ , and y ie lds
th iazo les unsubstitu ted in the 2 -position . It was hoped that trea tm en t
of th ioform am ide with chlorom ethyl vinyl ketone would y ie ld 4 -vinyl
th iazo le (XIII).
N CH=CH2
XIII
McMahon re p o r ts the sy n th esis of ethyl vinyl ketone by a
F r ie d e l-C ra f ts reac tio n of propionyl ch lo ride and ethylene, followed by
16the dehydrochlorination of the obtained ethyl /3-chloroethyl ketone
A pplication of th is m ethod using ch lo roace ty l ch lo ride to the p rep a ra tio n
of chlorom ethylyvinyl ketone, how ever, re su lte d in a black infusib le,
inso luble m a te r ia l, p resum ab ly som e s o r t of c ro s s - lin k e d polym er
(see ex p erim en ta l). Since m ethyl vinyl ketone is one of the m ost reac tiv e
17m onom ers known , it is possib le that po lym eriza tion o c c u rre d under
the reac tio n conditions em ployed.
Dodson and King developed a m ethod w hereby a th ioam ide and a
ketone a re heated with a halogen, th ereb y avoiding the n ecess ity of
18iso la ting the a -ha lo carbonyl , o r , a lte rn a te ly , d ispensing with the
halogen en tire ly , and substitu ting a s tro n g oxidizing agent, such as
19thionyl ch lo ride , su lfu ric acid , su lfu ry l ch lo ride , e tc . Both these
m ethods w ere a ttem pted with m ethyl vinyl ketone and th ioace tam ide ,
using iodine, thionyl ch lo ride , and su lfu ry l ch lo ride . A ll th re e reac tio n s

9
re su lte d only in unw orkable t a r s . P resu m ab ly , the rea c tiv ity of m ethyl
vinyl ketone m akes it unsu itab le fo r such rea c tio n s .
A ttem pts to syn thesize th ioacry lam ide by the p ro ced u res
rep o rted in the l i te ra tu re failed to y ield the d esired product. The
m ethod of F a irf ie ld , involving the addition of hydrogen su lfide to
20n itr i le s , when applied to a c ry lo n itr ile , y ielded a sa tu ra te d compound
which m ay be NC-CHgCH^SCHgCHg-CN. This compound contained
n itrogen and su lfu r, but the n. m . r . sp ec tru m showed a com plex m u lti
p le! at 7. 1‘T', and the in fra re d sp ec tru m showed strong n itr ile
-1 -1 absorp tion at 2250 cm and peaks in the reg ion 730-780 cm , strongly
21suggestive of the -C H g-S-C H g- linkage . P resu m ab ly , cyanoethylation
of hydrogen sulfide took p recedence
over th ioam ide fo rm ation . An a ttem pt to p re p a re th ioacry lam ide by
15E rle n m e y e r 's p ro ced u re fo r th ioform am ide re su lte d in an a lm ost
quantitative reco v ery of s ta r tin g m a te r ia l.
The d e s ired th ioacry lam ide and th iom ethacry lam ide w ere finally
acqu ired by a m odification of th is p ro ced u re (see experim en ta l).
CH2 = CH-CN + H2S ---- ^ NCCHgCHgSCHgCHgCN
50-60
CHC1
CH
CH2 = C - C - NH2
50-60°

10
The n .m . r . sp ec tru m of th ioacry lam ide showed vinyl abso rp tion at
4. 25 T and 3. TS'tf, as w ell as a sh a rp sing le t at 7. 84 'C', which was
assigned to the -SH proton on the b asis of com parison with the sp e c
tru m of th ioace tam ide . The n .m . r . sp ec tru m of th iom ethacry lam ide
showed vinyl abso rp tion at 4. 5 2 and 4 . 157f, the -SH sing le t at 7. 84
nf, and a m ethyl peak, sp lit into a doublet, at 8. 0 1 T . The in fra red
sp e c tra of both compounds showed the d isappearance of the carbonyl
bands. Both compounds had c o rre c t e lem en ta l an a ly ses . T h io ac ry la
m ide y ielded a D ie ls -A ld er adduct when tre a te d with cyclopentadiene
in refluxing ethanol, but th iom ethacry lam ide failed to r e a c t with
cyclopentadiene in anhydrous ethyl e th e r at room te m p e ra tu re o r in
refluxing ch lo ro fo rm o r ethanol (see experim en tal).
In o rd e r to obtain a vinyl th iazo le unsubstitu ted in both the
4- and 5- positions, it would be n e c e ssa ry to use a haloacetaldehyde.
W iley and England s ta te that it is p re fe ra b le to u se som e m ore stab le
22deriva tive of th ese . T here a re num erous accounts in the l i te ra tu re
of the use of a ce ta ls . ^ ^ In th is w ork, ch loroacetaldehyde d iethy la-
ce ta l as w ell as ch loroacetaldehyde was used .
T hioacry lam ide and ch lo roace ta l w ere heated with ethanol
until the ethanol evaporated . D issolving the res id u e in w a te r, t r e a t
m ent with sodium hydroxide, ex trac tio n w ith e th e r, and d is tilla tio n
re su lte d in a n early quantitative rec o v e ry of ch lo ro ace ta l. Since it
is n e c e ssa ry fo r the ace ta l to b reak down to the aldehyde before reac tio n
11
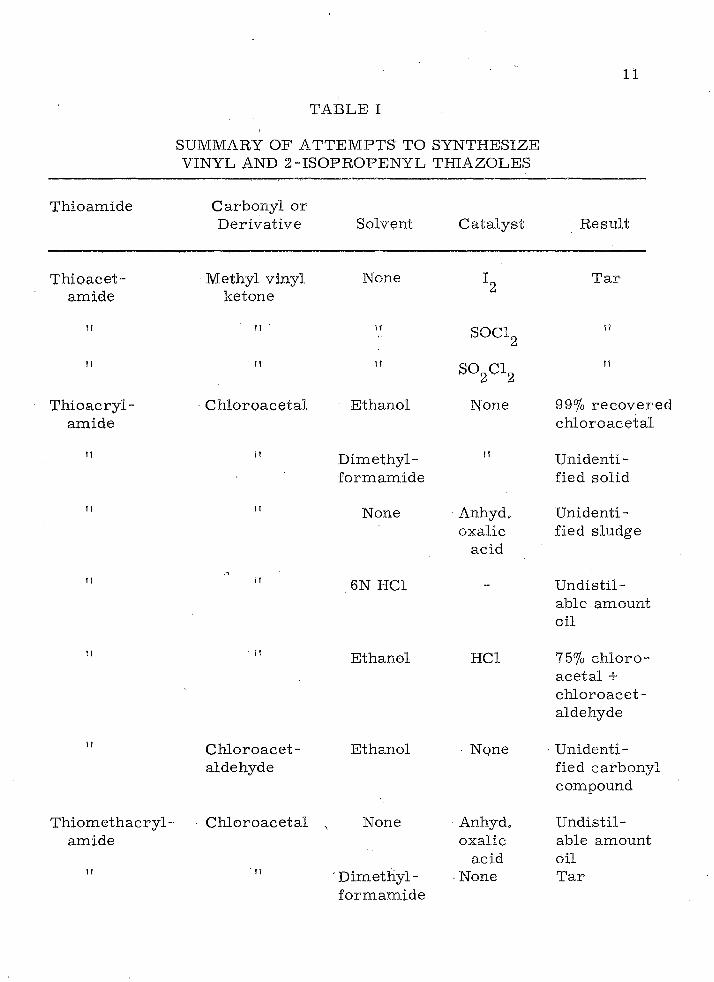
TABLE I
Thioam ide
SUMMARY OF ATTEM PTS TO SYNTHESIZE VINYL AND 2-ISOPROPENYL THIAZOLES
C arbonyl o r D erivative Solvent C ata lyst R esult
Thioacet-am ide
M ethyl vinyl ketone
None T ar
• M SOCL
T hioacry l-am ide
it
C h lo roaceta l E thanol
S02C12
None
D im ethyl- "fo rm am ide
99% rec o v e re d ch lo roace ta l
U nidentified solid
None Anhyd,oxalic
acid
U nidentified sludge
6N HC1 U ndistil- able am ount oil
E thanol
T hiom ethacryl-am ide
C hloroacet-aldehyde
Ethanol
C h lo roace ta l , None
" D im ettiyl-fo rm am ide
HC1 75% c h lo ro aceta l + ch lo ro ace t- aldehyde
None ■ U nidentified carbonyl compound
Anhyd.oxalic
acidNone
U ndistil- able amount oil T ar

12
can occu r, it is p robable that such decom position did not occu r under
under the reac tio n conditions. The fa ilu re to re c o v e r any th io a c ry la -
m ide can. be explained by the fac t th a t th is compound, is rea d ily soluble
in w a ter and. is , th e re fo re , lo s t in the w orkup.
An attem pted rea c tio n of the sam e two compounds, in refluxing
d im ethyIform am ide y ielded a white c ry s ta llin e so lid m elting a t 179-
180°. This compound contained ch lo rine , but n e ith e r n itro g en nor
su lfu r. The n. m . r . sp e c tru m failed to ind icate any v iny lic p ro tons,
and th is compound was not c h a ra c te r iz e d fu r th e r .
23The m ethod of H antzsch u tiliz e s anhydrous oxalic acid , p r e
sum ably to fac ilita te the decom position of ch lo ro ace ta l to ch lo ro ace ta l-
dehyde. The product obtained in th is case was a brow n sludge, which
could not be re c ry s ta l l iz e d o r iden tified .
When 6N hydroch lo ric acid w as em ployed as so lvent, the
re s id u a l o il a fte r rem ova l of the e th e r w as p re se n t in such sm all
am ounts that d is tilla tio n was not p o ss ib le . It is po ss ib le , but not
proven, tha t such a high acid concen tra tion did com pletely decom pose
the ace ta l, but that the aldehyde was lo s t in the w orkup. The fa ilu re of
th iazo le fo rm ation to occu r could not be explained a t th is point.
A. s im ila r rea c tio n of the th ioam ide and ch lo ro ace ta l in refluxing
ethanol with a tra c e of acid re su lte d in 75% rec o v e ry of u n reac ted
ch lo ro ace ta l, However, a sm a ll am ount of ch loroacetaldehyde was
obtained, indicating that som e h yd ro ly sis did o ccu r. The fac t that no

13
vinyl th iazo le was obtained seem ed .to suggest the p o ss ib ility of a
com petitive rea c tio n o ccu rrin g which would e ssen tia lly rem ove thp
th ioam ide from the rea c tio n . The obvious con jec tu re , of c o u rse , was
tha t the th ioam ide was reac tin g with, i ts e lf in som e way, A. study of
th iom ethacry lam ide showed what the n a tu re of th is rea c tio n was (see
below).
A m odification of the reac tio n , in troduced by H ro m a tk a ^ ^ ' ^/
avoids iso la tion of the th ioam ide, and co n sis ts of heating a m ix tu re of
the am ide, phosphorus pen tasu lfide , and the a -h a lo ca rb o n y l compound.
27A rea c tio n following the m ethod d esc rib ed by Schwarz fo r 2 ,4 -d i
m ethyl th iazo le , using aery lam id e , phosphorus pen tasu lfide , and
ch lo roacetone , fa iled to y ie ld the d e s ired product (2 - vinyl - 4 - m ethyl -
th iazo le). However, th is re fe re n c e m entions the p o ss ib ility of
obtaining a substitu ted oxazole in th is rea c tio n . T his e lim in a tes
hydro lysis of the th ioam ide as a rea so n fo r the fa ilu re of the p rev iously
d esc rib ed re a c tio n s , s ince , even w ere hyd ro ly sis to o ccu r, som e
2 -vinyl oxazole should be iso la ted , which w as not the case .
R eactions of th iom ethacry lam ide and ch lo ro ace ta l m et with no
m ore su c ce ss than did those with th io aery lam id e . In a ll c a se s , t a r s ,
rec o v ere d s ta r tin g m a te r ia l, o r frac tio n s too sm all to pu rify w ere the
re s u lt (see experim en ta l).
To de te rm ine the na tu re of a poss ib le com petitive reac tio n ,
th iom ethacry lam ide was re fluxed .in ethanol with a t r a c e of hydroch lo ric

14
acid in the absence of ch lo ro ace ta l. R em oval of the so lvent y ielded a
fo rm , carbon te tra c h lo rid e , acetone, and dioxane, but soluble in w ater,
m ethanol, ethanol, and d im ethyIsulfoxide. This m a te r ia l had an
inheren t v isco s ity of 0 .117 in d im ethylsulfoxide. If th is reac tio n takes
place rap id ly enough under the reac tio n conditions, it would explain the
fa ilu re to obtain the d e s ired th iazo les , since the th ioam ide is thus
e ssen tia lly rem oved from the reac tio n m ix tu re . Its so lub ility in w a ter
would cause it to be lo s t in the reac tio n workup, explaining the reco v ery
of only ch lo ro ace ta l.
Since it now appeared unfeasib le to u se these th ioam ides in the
sy n thesis of vinyl th iazo les , a tten tion was tu rned to the possib ility of
po lym erizing a, /3 -u n sa tu ra ted th ioam ides.
F re e rad ic a l po lym eriza tion would not be expected to yield high
m o lecu la r weight p o ly m ers, since the -SH group would ac t as a chain
t r a n s fe r agent.
Studying the b a se -ca ta ly zed po lym eriza tion of acry lam id e ,
28B reslow found that he obtained, not the expected sim ple vinyl po lm er,
but one which a ro se from a proton tr a n s fe r m echanism :
po lym eric white solid , m .p . > 250°, which was inso lub le in ch lo ro -
OCH 3
CH2 = CH -C-N H -CH 2 -CH -C-N H > CH2 = CH -C-N H -CH 2 -CH -C-N H

15
M arvel and Yoda obtained s im ila r re s u lts with jD -styrenesulfona-
29m ide. The polym er re su ltin g from the b a se -in itia te d po lym eriza tion
of th is m onom er proved to be one with m ixed re c u rr in g un its , som e
a ris in g from sim ple vinyl po lym eriza tion and o th ers fro m the proton
tr a n s fe r reac tio n , which would y ield a s tru c tu re (XIV). They assum ed
tha t the
SO N H—
XIV
p ro to n - tra n s fe r reac tio n was o ccu rrin g because of the d e c re a se in the
-NH- s tre tc h in g band at 3310 and 1615 cm *.
T h ioacry lam ide and th iom ethacry lam ide w ere po lym erized with
po tassium t-bu tox ide accord ing to M arv e l's p ro ced u re . They both
yielded white, powdery so lid s , m elting above 275°. The po lym er
obtained from th ioacry lam ide had an in heren t v isco sity of 0. 055, while
that of the th iom ethacry lam ide po lym er w as som ew hat h igher, 0 .171 .
The -NH- s tre tc h in the in fra re d sp ec tru m of po ly th ioacry lam ide
appears at the sam e w avelength as it does in the m onom er, 3325 and -1
3175 cm , while the po lym er obtained from th iom ethacry lam ide shows
the -NH- s tre tc h at som ew hat h igher w avelengths than does the m onom er,
-1 -1 3430 and 3180 cm as opposed to 3380 and 3175 cm

16
Two obvious s tru c tu re s a re possib le fo r the po lym er
n
SH
A B
B ecause of the deshielding effect of —C— and —NH— the pro tons bound to
carbon in s tru c tu re B would be expected to appear at considerab ly low er
fie lds than carbon bound protons in A.
The n. m . r . sp ec tru m of the th iom ethacry lam ide po lym er, while
poorly reso lv ed , shows no abso rp tion at low er fields which would in d i
cate pro ton t r a n s fe r . Two broad peaks, at 8. 75'X and 8. OSTf, having
re la tiv e peak a re a s of 3:2, seem to ind ica te that po lym eriza tion in th is
case o ccu rs p r im a rily , if not exclusively , through a sim ple vinyl
m echan ism , and that the po lym er has s tru c tu re A(R=CH^).
The n. m . r . sp ec tru m of the th ioacry lam ide po lym er, p resen ts
a m ore com plex p ic tu re . The absence of peaks in the reg ion lO'X to
8. 5 would seem to ind ica te that, in th is c ase , no sign ifican t amount
of vinyl po lym er is fo rm ed . The sp ec tru m shows peaks at 7. O S 't,
7. 21+t , 6. 9 0 f , and 6. 75 . It is possib le tha t the po lym er contains
se v e ra l types of re c u rr in g u n its . T hese can be postu lated read ily from
the m echan istic view, but a re difficult to a ss ig n to the sp ec tru m without
b road g en era liza tio n s .

17
One of these un its , of co u rse , is the one a ris in g from proton
t ra n s fe r , which would y ield a polym er of s tru c tu re (XV).
4-C H g -C H g -N H -f
(XV)
The prob lem with assign ing th is s tru c tu re is that the m ethylene
signals would be expected to appear as m u ltip le ts , w hereas the observed
signa ls a re apparen tly s in g le ts . Although the evidence is fa r from
32exhaustive, th e re is a p o ss ib ility that p ro tons a to C=S and £ to —NH—
fa ll in roughly the sam e a re a of the sp ec tru m as those in the re v e rse
c ase . The p robab ility of the two se ts of p ro tons having the sam e
chem ical sh ift, and appearing as a s ing le t is too unlikely to consider
se rio u s ly a t th is tim e .-1
The fact that the po lym er shows im ine abso rp tion at 1650 cm
in the in fra re d , which cannot be assigned to —C=NH as it could w ereSH
vinyl po lym er p re se n t, m akes possib le ano ther postu la tion concerning
the s tru c tu re of the po lym er, nam ely
-K:h2̂ h24 -̂6-^c
This s tru c tu re could a lso a r is e by the p ro ton tr a n s fe r m echanism ,
but involves a d ifferen t attack ing sp ec ie s than that req u ire d fo r B.
A ssum ing —SH to be the m ost acid ic sp e c ie s in the m onom er, th is
m echanism could be as follow s:
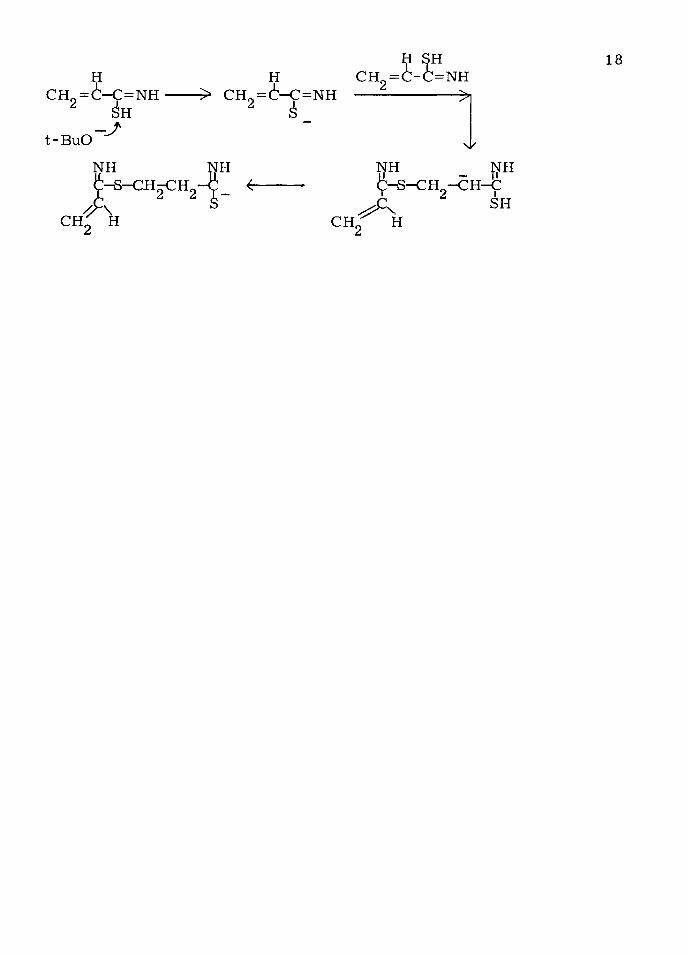
CH =6-C = N H
t-BuO
H-> c h = 6- c = n h
* &
NH NH
c n f h
=NH___
NH
18
CH
A -C -S -C H --C H -C^Cx sh_ H

EXPERIM ENTAL
M elting points a re unconnected. In fra re d sp e c tra w ere
de te rm ined on a P e rk in -E lm e r In fraco rd , c a lib ra ted against
po lysty rene; n. m . r . sp e c tra w ere de te rm ined on a V arian Model
A -60 (60 Me. )sp e c tro m e te r using te tram e th y ls ilan e as in te rn a l o r
ex te rn a l s tandard , M icroana ly ses w ere p e rfo rm ed by tbe M icro -
Tech L ab o ra to rie s , Skokie, Illino is. Solvents u sed w ere a ll reagen t
g rade and not red is tille d , w ith the exception of benzenp, which was
d ried over calcium ch lo ride and d is tilled , and d im ethy lfo rm am ide,
which w as d ried and d is tilled over calcium hydride. L iquid reag en ts
w ere d is tilled and so lid reag en ts re c ry s ta lliz e d p r io r to use w h erev e r
p o ss ib le . A nhydrous oxalic acid w as obtained by sub lim ation from
the d ihydrate . Hydroquinone w as added to a ll rea c tio n s in which
po ten tia lly po lym erizab le compounds w ere used ,
A. F r ie d e l-C ra f ts R eaction of C hloroaoety l C hloride and
16Ethylene
C hloroacety l ch lo ride w as p re p a re d from ch lo ro ace tic acid and
30thionyl ch loride by the m ethod of M cM aster and Ahwann. ' This
compound, boiling at 96-98°, showed carbonyl abso rp tion at 1790_ 1 ^
cm in the in fra red , ind icative of an acid ch lp ride .
19

C hloroacety l ch loride (4Qd g„ , 3. 54 m ole) was added slow ly to
a s t i r r e d m ix tu re of 2000 m l. of carbon disdlfide and 575 g. (4„ 3 m ole)
of anhydrous alum inum ch lo ride . The m ix tu re w as cooled to 0° w ith
s t i r r in g and d ry ethylene p assed into it fo r 7 hours, The m ix tu re
was heated on a s team bath at 40-45° un til b r isk evolution of HCl
dim inished and w as then allowed to stand at room te m p e ra tu re
overnight. The re s id u e w as poured onto ice to decom pose the
alum inum ch lo ride com plex, resu ltin g in a two phase sy stem . The
o rgan ic la y e r w as w ashed with dilute hyd roch lo ric acid, w a ter, and
dilute sodium bfcarbpnate un til an alkaline reac tio n w as obtained. .
The re su ltin g s ligh tly t a r r y solution w as d is tilled at a tm ospheric
p re s s u re to rem ove the so lvent. The re s id u e in the d is tillin g flask
was a black, infusib le , inso luble solid , w hich was not identified.
B. M ethyl Vinyl Ketone and T hioacetam ide w ith Sulfuryl
19C hloride
T hioacetam ide (36 g . , 0. 48 m ole) w as d isso lved in 14 g. (0 .2
m ole) of m ethyl vinyl ketone and the m ix tu re cooled in an ic e -s a lt
bath. Sulfuryl ch loride (27 g . , 0. 2 m ole) was added slow ly enough
to keep the exo therm ic rea c tio n to a m inim um . When addition was
com plete, the t a r r y m ix tu re was heated on a steam bath fo r 15
m inu tes. The re su lta n t ta r w as e x trac ted w ith e th e r, but e v ap o ra
tion of the e th e r y ielded only a: film on the inside of the flask .

21
C, M ethyl Vinyl Ketone and T hioacetam ide w ith Iodine '*'̂
T hioacetam ide (30 g„, 0„ 4 m ole) was d isso lved in 14 g„ (0. 2 m ole) of
m ethyl v inyl ketone and 50. 8 g. (0. 2 m ole) of iodine added portionw ise
th rough a re flux condenser. The rea c tio n w as highly exo therm ic , and
gave off dense white fum es. When addition w as com plete and the r e
action subsided , the m ix tu re w as refluxed gently fo r 2 hours. The
resu ltin g b lack ta r was diluted, w ith w ater and heated un til m axim um
solution o c cu rred . The aqueous solution w as m ade b asic w ith con
cen tra ted am m onium hydroxide and e x trac ted w ith anhydrous e ther.
The com bined e th e rea l e x tra c ts w ere d ried oversigh t over solid
sodium hydroxide, and the e th e r rem oved under vacuum . A ttem pts
to d is till the re s id u a l o il re su lte d in the fo rm ation of a b lack so lid in
the d istilling flask .
D. M ethyl Vinyl Ketone and T hioacetam ide w ith Thionyl
C h lo rid e '*' ̂
T hioacetam ide (3 0 g ., 0. 4 m ole) and 14 g. (0. 2 m ole) of m ethyl
vinyl ketone in a D ry Ice - acetone bath w ere tre a te d 11.9 g. (0.1
m ole) of thionyl ch lo ride . The resu ltin g b lack ta r w as w orked up
in the m anner d esc rib ed above (see C) w ith the sam e re s u lt.
20E. A cry lp n itrile and Hydrogen Sulfide
A cry lo n itrile (222 g. , 4. 2 m ole) w as d isso lved in 232 m l. of
pyrid ine and 101 g. (1. 0 m ole) of tr ie th y lam in e w as added. Hydrogen

22
sulfide was bubbled through the m ix tu re in a steady s tre a m for 3. 5
hours. The m ix tu re w as poured into d is tilled w a te r and the organic
la y e r rem oved. The aqueous la y e r w as ex trac ted w ith e th e r. The
com bined organic frac tio n s w ere d is tilled to rem ove so lven ts, and
the re s id u a l v ile -sm e llin g red oil cooled in a D ry Ice -acetone bath
in an e ffo rt to p rec ip ita te the suspended c ry s ta ls . F iltra tio n and
re c ry s ta ll iz a tio n from ethyl ace ta te y ielded 4. 0 g. of a pale yellow
solid, m . p. 35-36°. The in fra re d sp ec tru m of th is so lid showed-1
strong n itr ile absorp tion at 2250 cm and peaks in the reg ion 730 -
-1 21 780 cm , strong ly suggestive of the -C H ^-S-C H g- linkage.
F u rth e rm o re , the n. m . r . sp ec tru m showed no vinyl p ro tons, a com
plex m ultip le! at 7. 1 T , and s ing le ts at 8. 0 and 9. 55MT , both of
which a re p robably caused by im p u ritie s . This compound was
assum ed to be NCCH^CH^SCH^CH^CN, a ris in g from the cyanoethy-
la tion of hydrogen sulfide.
15F. A crylam ide and P hosphorus P en tasu lfide in Xylene
To 71 g. (1 m ole) of acry lam ide suspended in xylene in an ice -
sa lt bath w as added 35 g. (0. 16 m ole) of phosphorus pen tasulfide
w ith v igorous s t i r r in g over a period of two hours. The m ix tu re was
then left overnight w ith s t ir r in g . The xylene was se p a ra te d from the
re s id u a l solid and the so lid ex trac ted w ith e ther. C oncentration of
the com bined organ ic la y e rs and the addition of pe tro leum e th e r

23
(30-60°) re su lte d in the p rec ip ita tio n of a white solid , which, a fte r
re c ry s ta lliz a tio n from ethyl ace ta te , was shown to be u n reac ted
s ta r tin g m a te r ia l on the b a s is of in fra re d spectrum and m ixed
m elting point. T otal y ield of u n reac ted s ta r tin g m a te r ia l - 65 g.
(93%).
G. A crylam ide and Phosphorus P en tasu lfide in C hloroform
A cry lam ide (200 g. , 2. 82 m ole) w as d isso lved in 400 m l. of
ch lo ro fo rm and 100 g. (0. 45 m ole) of phosphorus pen tasu lfide added
w ith s t ir r in g at 50-60° over a pe riod of 2 hours. The m ix tu re was
s t i r r e d w ithout heating overn ight and then allowed to stand without
s t i r r in g fo r 48 hours. The re su ltin g tw o -lay e r sy s tem was ex trac ted
with anhydrous e th e r and the e th e r concen tra ted on a s team bath
under a sp ira to r vacuum . On the addition of pe tro leum e th e r (30-60°)
a white so lid p rec ip ita ted . Two re c ry s ta ll iz a tio n s from ethyl ace ta te
yielded 6 g. (15.3%), m. p. 67-68°. (R epetition of the p rocedu re
eventually in c re a sed the yield to 18 g. (45. 9%) ). The n. m . r . sp e c
tru m showed vinyl absorp tion at 4. 25 ~C and 3. 75‘C and a sharp
sing let at 7. 84 ■T which w as assigned to -SH on the b a s is of com
p a riso n w ith the sp ec tru m of th ioace tam ide . The in fra re d spectrum
showed the d isappearance of the carbonyl band.
Anal. C alcd. fo r C^H^NS: C, 41. 34; H, 5. 78; N, 16.07.
Found: C, 41. 38; H 5. 97: N, 15. 95

24
EL M ethacry lam ide and P hosphorus P en tasu lfide in C hloroform
M ethacrylam ide (200 g„ , 2. 38 m ole) w as d isso lved in 400 m l. of
ch lo ro fo rm (gentle heating) and w as 'tre a te d with 100 g. (.0. 45 mole)
of phosphorus pen tasu lfide in the m anner d escrib ed in (G r). Workup
w as the sa tne except that the reac tio n m ix tu re did not stand, fo r 48
hours a fte r overnight s t ir r in g . The y ield of w hite, pow dery solid , m. p.
101-102° a fte r re c ry s ta ll iz a tio n from ethyl ace ta te w as 20 g. (44%).
A nal: C alcd. fo r C ^ N S : C, 4-7. 50; H, 6. 83; N, 13.85.
Found: C, 48.01; H, 7. 15 ;N , 13. 73.
22I. T h ioacry lam ide and C hloroacetaldehyde
T hioacry lam ide (5 g . , ,0. 057 m ole) w as d isso lved in 75 m l. 95%
ethanol and 4. 5 g. (0. 057 m ole) ch loroacetaldehyde added. The m ix tu re
w as then refluxed fo r 8. 5 hours, a fte r which it w as poured into w ater,
m ade b asic w ith sodium hydroxide, and ex trac ted w ith anhydrous e th e r.
The e th e r e x tra c ts w ere d ried , and the e th e r rem oved under vacuum .
D istilla tion of the re s id u a l red d ish brow n oil yielded 2 m l, of a c le a r,
c o lo rle ss liquid, b. p. 25° (0. 5m m .). The n. m . r . sp ec tru m showed
no vinyl p ro tons, and the in fra re d sp ec tru m showed a s tro n g carbonyl -1
band at 1750 cm . The product contained no su lfu r o r n itrogen , but
did contain ch lo rine . This product w as not fu rth e r identified .

25
J , T hioacry lam ide and C M oroacetal in E thanol
T h ioacry lam ide (34. 8 g. t 0. 4 m ole) and 61. 04 g. (0. 4 m ole) of
ch lo ro ace ta l w ere heated on a steam bath in 175 m l. 95% ethanol
until the ethanol evaporated . The re s id u e w as w orked up in the u sua l
m anner (see I..). D istilla tion of the re s id u a l liquid y ielded 50 g. of
a c le a r , c o lo rle ss liquid, b. p. 44-47° ( 5. 5mm) which contained no
su lfu r o r n itrogen , but did contain ch lorine; The in fra re d sp ec tru m
of th is m a te r ia l w as superim posab le on tha t of ch lo roace ta l.
27K. A cry lam ide, Phosphorus P en tasu lfid e , and C hloroacetone
A m ix tu re of 150 g. (2. 11 m ole) and 100 g. (0. 45 m ole) of phor-
phorus pentasu lfide w as added to 100 m l. of dry benzene. To th is
w as added 20 m l. of a m ix tu re of 200 m l. of ch loroacetone in 75 m l,
of d ry benzene. The flask w as heated on a w a te r bath to in itia te the
reac tion , a fte r which the bath w as rem oved and the re m a in d e r of the
ch lo roacetone-benzene added portionw ise through a re flu x condenser.
A fter about o n e -th ird of the m ix tu re had been added, a so lid form ed
in the flask , m aking s t i r r in g im possib le and n ece ss ita tin g shaking
the flask a fte r each addition in o rd e r to obtain at le a s t p a r tia l m ixing.
When all of the ch lo roacetone-benzene m ix tu re had been added, the
flask, was heated on a steam bath fo r 1 /2 hour and .375 m l. of w ater
was added. The rea c tio n m ix tu re w as then allowed to stand at room
tem p e ra tu re fo r 2 hours. A fter th is tim e, the tw o-phase sy stem was

26
sep a ra ted . The low er la y e r , a deep re d oil, w as m ade.basic-w ith
po tassium hydroxide and ex trac ted w ith e th e r. The e th e r e x tra c ts
w ere d ried , f ilte re d , and the e th e r rem oved. D istilla tio n yielded
5 g. of a s ligh tly v iscous orange liquid, b„ p. 76-83° (2-2 . 5mm), th e .
in fra re d sp ec tru m of which showed strong carbonyl absorp tion at -1
1720 cm and whose n. m . r . sp ec tru m indicated the absence of
a rom atic p ro tons.
2 5L. T hioacry lam ide and C h lo roaceta l in H ydrochloric Acid
T hioacry lam ide (17. 8 g . , 0. 204 m ole) and 30. 8 g. (0. 204 mole)
of ch lo ro ace ta l w ere refluxed Under n itrogen in 100 m l, 6N hydro -
ch lo ric acid for 1 hour. The dark, v iscous m ix tu re w as w orked up
in the u su a l m anner (see I . ), and, a f te r evaporation of the e ther,
th e re rem ained on ly a.few drops Of a brow n oil - not enough to d is till.
23M. T hioacry lam ide and C h lo roaceta l w ith Anhydrous O xalic Acid
A m ix tu re of 19. 4 g. (0. 127 m ole) of ch lo ro ace ta l and 11.4 g.
(0.127 m ole) of anhydrous oxalic acid w as heated under a reflux
ocondenser in an oil ba th at 140 un til gas evolution ceased . To the
solution w as then added 11.0 g; (0. 127 m ole) of th io acry lam id e .
Gentle heating in itia ted a b r is k reac tio n , w hereupon heat w as rem oved
and the reac tio n allowed to p roceed un til it subsided (about 1/2 hour),
oThe d ark m ix tu re w as then heated fo r an additional hour at 100 ,
allowed to cool, and diluted w ith 100 m l. of dilute hydroch lo ric acid

27
and 100 m l. of w a te r. F iltra tio n rem oved the p rec ip ita ted so lid s,
and the c le a r , d a rk f i ltra te w as m ade b asic w ith 5N po tassium
hydroxide and ex trac ted w ith e th e r. An attem pt to d is till the r e s i
dual d a rk oil under vacuum a fte r rem ova l of the e th e r re su lte d in
the fo rm ation pf a so lid in the d is tilla tio n flask (bath tem p era tu re
< 100°). T his solid , d ifficu lty soluble in m ost o rgan ic so lven ts,
could not be re c ry s ta ll iz e d , rem ain ing a brow n sludge a fte r all
a ttem pts.
N. T h ioacry lam ide and C h lo roaceta l in Ethanol (Acid Catalyzed)
A m ix tu re of 8. 7 g. (0. 1 m ole) of th ioacry lam ide , 15. 3 g. (0, 1
m ole) of ch lo roace ta l, and 3 ml. of concen tra ted hydroch lo ric acid
was refluxed in .95% ethanol fp r 12 h ou rs. The dark m ix tu re was
allowed to cool and w orked up in the u su a l m anner (see I , ). D is til
lation of the re s id u a l d a rk liquid under a sp ira to r vacuum yielded
3. 5 g. of ch loroacetaldehyde and 11. 5 g. of ch lo ro ace ta l (identified
by its in fra re d spectrum ),
O. . T h iom ethacry lam ide and C h lo roaceta l w ith A nhydrous
Oxalic Acid
The p rocedu re d esc rib ed in (M .) w as followed, using 10,1 g,
(0.1 m ole) of th iom ethacry lam ide , 15. 3 g. (.0.1 m ole) of ch lo roace ta l,
and 9. 0 g. (0. 1 m ole) of anhydrous oxalic acid. The re s id u e re m a in
ing a fte r rem oval of the e th e r w as not enough to d is till, .even" in
m ic ro -a p p a ra tu s .

28
P. Thiom ethacryXam ide and C h lo race ta l in D im ethylform am ide
T hiom ethacry lam ide (10. 1 g . , 0. 1 m ole) and 15. 3 g„ (0. 1 mole)
of ch lo roace ta l in 50 m l. of d im ethylform am ide w ere heated with
os t ir r in g in a w a ter ba th at 95 fo r 4 h ou rs. When the heating was
com pleted, the dark brow n m ix tu re w as w orked up as d esc rib ed
above (see I. ), A ttem pt to d is till the re s id u a l oil re su lte d in the
fo rm ation Of an und istillab le t a r in the d is tilla tio n flask .
Q. T hioacry lam ide and Cyclopentadiene
A m ix tu re of 8. 7 g. (.0. 1 m ole) of th ioacry lam ide and 6. 6 g.
(0. 1 m ole) of cyclopentadiene (d is tilled im m ed ia te ly befo re use
through a 12-inch colum n packed w ith g la ss he lices and caught in a
D ry Ice-coo led receiving, flask) w as refluxed in 100% ethanol fo r 6
hours. Rem oval of the so lvent yielded 7. 4 g. of a white solid , m, p.
198-199°. Its low so lub ility in m ost o rgan ic so lven ts m ade c h a ra c
te r iz a tio n difficult, but the n. m . r . sp ec tru m showed the d isappearance
of the vinyl p ro tons of th ioacry lam ide , and the appearance of h igher
33field peaks c h a ra c te r is t ic of the bicycloheptene sy stem , although
the low field peaks appear to be absent.
R. T hiom ethacry lam ide and Cyclopentadiene
M ixtures of 12, 1 g. (0. 12 m ole) of th iom ethacry lam ide and 7. 92 g.
(0, 12 m ole) of f re sh ly d is tilled cyclopentadiene w ere tre a te d as follow s:
1, In anhydrous e th e r at room te m p e ra tu re fo r 12 hou rs.

29
2. In refluxing ch lo ro fo rm fo r 8 hours.
3. In refluxing absolute ethanol fo r 18 hours.
All reac tio n s re su lte d in an e sse n tia lly quantitative re c o v e ry of
s ta r tin g m a te ria l, as de te rm ined by sp e c tra and m ixed m elting point.
S. T h iom ethacry lam ide w ith H ydrochloric Acid in Ethanol
A m ix tu re of 5 .0 g. (0 .05 m ole) of th iom ethacry lam ide , 2 m l. of
concen tra ted hydroch lo ric acid, and 50 m l. of 95% ethanol was r e -
fluxed fo r 6 hours. Rem oval of the g re a te r p a rt of the solvent on a
ro ta ry ev ap o ra to r y ielded 3. 8 g. of a white solid . The crude
m a te r ia l, m . p. > 250°, was insoluble in ch loroform , carbon t e t r a
ch loride, acetone, and dioxane, and soluble in w ater, m ethanol,
ethanol, and dim ethylsulfoxide. The so lid was d isso lved in d im ethy l-
sulfoxide and poured into ch lo ro fo rm , resu ltin g in the p rec ip ita tion
of a fine, white pow der, w hich was f ilte re d and d ried in vacuo at 70°
fo r 12 hours. rCj (0. 5% in DMSO) 1. 06; ^ 0. 117.
29T. B ase-C ata lyzed P o ly m eriza tio n of T hioacry lam ide
P o ta ss iu m t-bu tox ide w as p rep a red by adding 39 g. of fre sh ly
cut po tassium to 74. 1 g. of d ry t-b u ty l alcohol (d ried and d is tilled
over sodium ) and refluxing the m ix tu re fo r 15 hours. The excess
t-b u ty l alcohol w as d is tilled under reduced p re s su re , and the product
was obtained as a white pow der free from the alcohol.

30
A solution of 3. 87 g. of th ioacry lam ide in 20 m l. of d ry d im ethy l-
fo rm am ide in a 100-m l 3-necked flask equipped with a m agnetic s t i r r e r
and reflux condenser w as heated w ith 0. 00 54 g. of phenyl- (3 -naphthala-
quickly added 0.0598 g. of po tassium t-bu tox ide, and the m ix ture
s t i r r e d at the sam e te m p e ra tu re fo r 30 hours. Solid m a te r ia l was
noted in the flask a fte r about 10 hours.
The reac tio n was quenched by pouring into w a ter containing 10%
ethanol and a tra c e of hydroch lo ric acid. Much of the solid m a te ria l
d isso lved , so the aqueous m ix tu re w as poured into m ethanol, r e s u l t
ing in a m ore copious p rec ip ita te . The so lid was f ilte re d and dried
in vacuo at 70° fo r 12 hours, yielding 2. 5 g. of a white powder, m. p.
U. B ase-C ata lyzed P o ly m eriza tio n of T h iom ethacry lam ide
The sam e m ethod d esc rib ed above, using 4. 38 g. of th io m e th acry
lam ide, 0 .0027 g. of phenyl-j3 -naphthalam ine, and 0.0293 g. of
po tassium t-bu tox ide. The reac tio n m ix tu re was heated under helium
for 36 hours, so lid being noted a fte r about 8 hours.
E x trac tion of the f iltra te w ith e th e r and evaporation yielded 0. 8 g.
of u n reac ted th iom ethacry lam ide . T otal y ield of po lym er a fte r drying:
3. 0 g. of white pow der, m . p. > 275°. rn r e ̂ (0. 0 5% in DMSO) 1. 009;
m ine at 135° under n itrogen fo r 1 hour. To th is m ix tu re w as then
> 275 o (0. 05% in DMSO)re l

REFERENCES
1. M. M. Koton, J . P o ly m er SgL , 30, 331 (1958)
2. . C. W alling, E. R. B riggs, and K„ W olfstirn , J . Am. Chem.. S o c ., 70, 1543 (1948)
3„ . R„ L. F ra n k et al, Ind, Eng, C h e m ., 40, 879 (1948)
4. R. L. F ra n k et al, Ind. Eng. C h e m ., 40, 420 (1948)
5. S. KamenS'r, I. Sim ek, and E. R egensbogenour, Chem . Z v e s ti . , 14, 581 (1960); see Chem . A b s tr . , 55, 15450 i (1961)
6. E. J . M eehan, J . P o ly m e r-S e i,, 1̂ , 175 (1946)
7. E .'T . B orrow s et al, J.. Appl. Chem,. (London),h, 379 (1955)
8. . G. E, Ham, "C opolym erization", H igh-Polym ers, Vol. XVIII,In te rsc ien ce P u b lish e rs , New Y ork (1964), p. 509
9. C. E. Schildknecht, "Vinyl and R elated P o ly m ers" , John W iley & Sons, I n c . , New Y ork (1952), pp. 667-674
10. C. G. O y e rb e rg e r and F . W, M ichelotti, J . Am. Chem . Soc.,80, 988 (1958)
11. E, R. Buchm an and E. M. R ichardson, J . Am. Chem . S o c .,67, 395 (1945)
12. D. L. Schoene, J.. Am. Chem . S o c ., 73, 1970 (1951)
13. U nited .S tates Rubber C o ., B rit. P a t. 609, 467; see Chem .A b s tr . , 43; P 2805e
14. F o r a m ore deta iled d iscussion of th is reac tion , see R, H. W iley and B .C . E ngland,"O rganic R eactions" Vol. VI, John W iley & Sons, In c ., New Y ork (1951)
15. H. E rle n m e y er and K. M enzl, Helv. Chim, Acta, 31_, 2071 (1948)
31

32
16, E. M, McMahon et al, J„ Am, C h e m . - S g c , , 70, 2971 (1948)
17. Ref, 9, p. 687
18, R, M„ . Dodson and L. C„ King, J . Am. Chem,. S o c ., 67, 2242.(1945)
19. :R„ M„ Dodson and L. C. King, J . Am. Chem . S o c ., 68, 871 (194.6)
2.0. • "A. E . F a irfie ld , J . L. Lowe, and D; A. Peake, J , Chem . S o c ,,742 (1952)
21, R. ]V|. S ilv e rs te in and G. C. B a ss le r , l,S pec tropho tom etricIdentification of O rganic Com pounds11, John W iley & Sons, In c . , New Y ork (1964) p. 57
22.. Ref. 14, p. 378
2.3.. A . H antzsch, A n n ., 250, 257 (1889)
24. V. Roubleff, A n n ., 259, 253 (1890)
25. G. E. H. S k rim sh ire , Brit.- P a t. 540, 032; see Chem A b s t r . , 36, 4138 (1942)
2:6. a. O, H rom atka, U .S . P a t. 2, 160, 867; see Chem.. A b s t r . , 33, 7320 (1939)
b. O. H rom atka, G er. P a t. 670, 131; see Chem. A b s t r . , 33, 2909 (1939)
27. G. Schw arz,"O rganic Synthesis^ Coll. Vol. Ill, p. 332:,. John W iley and Sons, In c ., New Y ork (1955)
28. D. S. B reslow , G. OB. H ulse, and. A. S. M atlack, J . Am.. Chem, S o c ., 79, 3760 (1957)
29. N. Yoda and C. S. M arvel, J . P o ly m er S c i . ,. A_3, 2229 (1965)
30. L. M cM aster and. F . F . Ahmann, J . Am. Chem. S o c ., _50, 145:(1928)
31. Ref. 21, p. 62
32. Ref. 21, pp.. 83-4
33.. V arian A sso c ia tes , "NMR S p ec tra C atalogue", Vol. II