assessment of a biogas-generating microbial community in a pilot-scale anaerobic reactor
TRANSCRIPT

www.elsevier.com/locate/jbiosc
Journal of Bioscience and BioengineeringVOL. 117 No. 6, 730e736, 2014
Assessment of a biogas-generating microbial community in a pilot-scale anaerobicreactor
Elvira E. Ziganshina, Alsu R. Bagmanova, Irina V. Khilyas, and Ayrat M. Ziganshin*
Department of Microbiology, Kazan (Volga Region) Federal University, Kremlyovskaya str. 18, 420008 Kazan, The Republic of Tatarstan, Russia
Received 19 July 2013; accepted 18 November 2013Available online 10 January 2014
* CorrespondE-mail add
1389-1723/$http://dx.doi
In this work bacteria and methanogenic archaea utilizing agricultural wastes in a pilot-scale biogas reactor wereexamined using sequencing and terminal restriction fragment length polymorphism analysis. Based on the analyses of16S rRNA genes, Clostridia represented the most diverse group in the digester. Of the Clostridia, unclassified Clostridialesand the members of the genera Anaerotruncus and Tissierella were detected at high abundances. The representatives ofthe bacterial phyla Bacteroidetes and Proteobacteria were also defined, but in minor proportions, and were assigned tonon-dominant communities. Within the phylum Euryarchaeota, the members of the orders Methanosarcinales andMethanomicrobiales were found at high levels. Methanogenic archaea were analyzed using both 16S rRNA and mcrAgenes. Actually good results were received using both approaches; however, the rRNA gene method missed the non-dominant order Methanobacteriales.
� 2013, The Society for Biotechnology, Japan. All rights reserved.
[Key words: Biogas; Agricultural wastes; 16S rRNA genes; mcrA genes; Terminal restriction fragment length polymorphism]
Many research works have been devoted to the development ofnew clean and renewable alternative energy sources due to theincrease of gas and oil prices, the depletion of energy resources, andthe necessity to protect the environment from global warming. Oneof the perspective alternative energy sources is biomass. Largeamounts of organic wastes, such as agricultural and municipalwastes, become valuable sources of energy. In this connection, it isnecessary to create new processing and recycling technologies,including biotechnology for microbial conversion of organic resi-dues with biogas production, which is considered as one of themost efficient and environmentally attractive methods (1e3).
Anaerobic treatment of different organic wastes by microbes,involving the members of the Bacteria and Euryarchaeota, appearsto be the effective biotechnological method to convert biowastesinto bioenergy. Intensive process of anaerobic digestion of bio-wastes requires the maintenance of certain optimal microbialprocesses. As a consequence, many researchers have focused theirworks on the production of biogas from biodegradable materials(e.g., agricultural, municipal, and industrial wastes as well assewage sludge) and have made successful efforts on the investi-gation of the structure and dynamics of biogas-producingmicrobialassociations in various bioreactors (2,4e10).
Except for the cultured collection of bacterial and methanogenicstrains, in the last years a large group of uncultured microorgan-isms was discovered. Since it is difficult to study anaerobic micro-organisms with culture-based methods, culture-independentmolecular methods were intensively developed to investigatecomplex bacterial and archaeal communities in the environment.
ing author. Tel.: þ7 843 2337872; fax: þ7 843 2387121.ress: [email protected] (A.M. Ziganshin).
e see front matter � 2013, The Society for Biotechnology, Japan..org/10.1016/j.jbiosc.2013.11.013
These methods allow the studying prokaryotic communities di-versity based on 16S rRNA gene and some functional genes.Methanogens can be effectively analyzed based on a subunit ofmethyl coenzyme M reductase (mcrA) gene, molecular metabolicmarker of methanogenesis (11e14). Methyl coenzymeM reductase,which is peculiar to methanogenic archaea, catalyzes the reductionof methyl coenzyme M with coenzyme B to heterodisulfide andmethane under anaerobic conditions (15). Molecular biologytechniques developed for the characterization of microbial pop-ulations, which carry out the anaerobic treatment of biomass, allowresearchers to monitor microbial interactions and ultimately willhelp to improve the efficiency of the whole anaerobic digestionprocess.
Within the main research topics, to achieve a more stableanaerobic digestion process and to avoid its failure, should be theselection of well-adapted microbial populations based on thesubstrate composition as well as the investigation of the keybiochemical pathways for various organic compounds degradationwith biogas production. Scientific advances in the establishment ofhighly active and well-adapted members of biogas-producing mi-crobial communities should be a breakthrough in solving theproblem of biowastes disposal and, at the same time, should pro-vide a scientific basis for regulation of the anaerobic process withbiogas generation. Recently we investigated bacteria and archaeainvolved in anaerobic digestion of multifarious organic waste ma-terials in lab-scale digesters (10,16), and in this research we set agoal to determine bacterial and methanogenic archaeal communitydiversity in a pilot-scale biogas reactor. The feedstock for thebioreactor was composed primarily of cattle manure and plantbiomass. The diversity of the bacterial community involved in thisanaerobic process was assessed by creating clone libraries for thebacterial 16S rRNA genes and by terminal restriction fragment
All rights reserved.

VOL. 117, 2014 BIOGAS-GENERATING MICROBIAL COMMUNITY 731
length polymorphism (T-RFLP) analysis. For methanogenic archaeacomposition, the community structure was analyzed using both16S rRNA and mcrA genes.
MATERIALS AND METHODS
Operation of the biogas reactor and analytical techniques The agricul-tural biogas reactor is located in a dairy farm in the Buinsky municipal district, theRepublic of Tatarstan, Russia. This is the first pilot-scale continuous stirred tankreactor constructed in the Republic of Tatarstan in 2010. The anaerobic digester witha working volume of 25 m3 was continuously fed with agricultural wastes, primarilywith cattle manure and plant biomass, and operated stably at mesophilic regime(38e39�C). The reactor had been running stably for more than 6 months when theresearch was started. Samples for analyses were taken in October, 2011 and March,2012 and then analyzed as described in our recent publications (10,16). The biogasyield varied in the range of 304e331 L kg�1 volatile solids and the methanecontent was in the range of 52e55% during the reactor operation. The pH valuewas kept at 7.6e7.8 in the digester. The concentrations of organic acids andammonium in the first sample achieved 1.1 � 0.06 g L�1 and 0.71 � 0.02 g L�1
values, respectively, while the organic acids and ammonium amounts in thesecond sample were 1.3 � 0.08 g L�1 and 0.85 � 0.03 g L�1, accordingly.
DNA extraction and purification Samples for microbial community ana-lyses were collected into sterile 15 mL Falcon tubes and used immediately for DNAextraction and purification. DNAwas eluted and purified from 0.25 mL of the samplewith a PowerSoil DNA Isolation Kit (MO BIO, USA), checked by 1.5% agarose gelelectrophoresis, and quantified with a NanoVue Plus UVeVis spectrophotometer(GE Healthcare, USA).
16S rRNA and mcrA genes amplification, cloning, and sequencing Bacterial16S rRNA gene fragments were PCR-amplified using the primers UniBac27F (50-GAGTTT GAT CMT GGC TCA G-30) and Univ1492R (50-TAC GGY TAC CTT GTT ACG ACT T-30),whereas archaeal 16S rRNA gene fragments were amplified with the forward primerUniArc21F (50-TTC YGK TTGATC CYG SCRG-30) and the reverse primerUniArc931R (50-CCC GCC AAT TCC TTT HAG-30) as described previously by us (10,16).McrA genes wereamplified by using a combination of the primers mcrA-mlas (50-GGT GGT GTM GGDTTC ACM CAR TA-30) and mcrA-rev (50-CGT TCA TBG CGT AGT TVG GRT AGT-30) asdescribed by Steinberg and Regan (12).
Amplified 16S rRNA and mcrA genes were checked in 1.5% gel electrophoresisand then purified using a QIAquick PCR Purification Kit (Qiagen, Germany). Puri-fied genes were cloned using InsT/Aclone PCR Cloning Kit (Fermentas, Lithuania)according to manufacturer’s recommendations. The presence of the appropriateinserts of bacterial and archaeal 16S rRNA genes as well as mcrA genes in positiveclones was analyzed using the vector-specific M13 primers. 1 ml of the obtainedamplicons were further treated with the restriction enzyme HaeIII (New EnglandBiolabs, Germany) and separated electrophoretically. The restriction patternswere analyzed using a Phoretix 1D software (Nonlinear Dynamics, UK). Repre-sentative clones from all clusters were chosen for partial 16S rRNA or mcrAsequencing, which was performed in Syntol Labs (Moscow). The received 16SrRNA data were compared to the NCBI database by using the BLASTN program(http://www.ncbi.nlm.nih.gov/BLAST) and taxonomically assigned in appliancewith the RDP Classifier (http://rdp.cme.msu.edu). McrA sequences were analyzedusing BLASTX program (http://www.ncbi.nlm.nih.gov/BLAST). Data were checkedfor chimeric sequences with Bellerophon (http://comp-bio.anu.edu.au/bellerophon/bellerophon.pl). The partial sequences of 16S rRNA and mcrA genesreceived in this work were deposited in the GenBank database (accession numbersKF419189eKF419207).
T-RFLP analysis T-RFLP analysis was performed in accordance with ourprevious works (10,16). Bacterial 16S rRNA genes were PCR-amplified withUniBac27F-FAM and Univ1492R, archaeal 16S rRNA genes using the primersUniArc21F-FAM and UniArc931R, and mcrA genes with the primers mcrA-mlasand mcrA-rev-FAM. Amplicons were then cleaned up with a QIAquick PCRPurification Kit (Qiagen, Germany). Bacterial 16S rRNA genes were then subject to
TABLE 1. Sequencing results of representative bacterial 16S rRNA gene clone
OTU Clone (bp) Acc. no. Closest affiliationa (acc. no.)/% sim
OTU 1 bac_B1 (322) KF419195 Thermoanaerobacter uzonensis JW/IW_A615OTU 2 bac_D5 (425) KF419199 Desulfotomaculum kuznetsovii strain 17 (AY0OTU 3 bac_G4 (590) KF419202 Clostridium sp. 6-31 (FJ808611)/86%
bac_D3 (403) KF419198 Clostridium sp. 6-31 (FJ808611)/87%OTU 4 bac_A5 (471) KF419194 Anaerotruncus colihominis strain S6 (KC2060OTU 5 bac_B5 (542) KF419196 Tissierella creatinophila strain Kre4 (NR_0370OTU 6 bac_A4 (544) KF419193 Rumen bacterium NK4A65 (GU324373)/92%OTU 7 bac_F4 (636) KF419201 Flexibacter aggregans strain: IFO 15974 (AB0OTU 8 bac_E1 (541) KF419200 Bacteroidales bacterium RM68 (AB730709)/8OTU 9 bac_C4 (602) KF419197 Castellaniella sp. MJ05 (GQ250433)/97%
a Uncultured/environmental sample sequences were excluded from the analyses.
restriction enzyme digestion with the restriction endonucleases HaeIII and RsaI(New England Biolabs), archaeal 16S rRNA genes were cut with HaeIII and MseI(New England Biolabs), and mcrA genes were digested with HaeIII and MspI (NewEngland Biolabs).
GeneScan 500 ROX and GeneScan 1200 LIZ Size Standards (Applied Bio-systems, USA) were used to obtain the molecular size of restriction patterns.Fluorescently labeled T-RFs were sized on a genetic analyzer in Syntol Labs, and T-RFLP fingerprint patterns were analyzed using Peak Scanner Software v1.0(Applied Biosystems). T-RFs of archaeal 16S rRNA genes that were <35 bp and>900 bp in size were excluded from further analyses; T-RFs of bacterial 16S rRNAgenes and mcrA genes <50 bp and >500 bp were also removed from subsequentanalyses. T-RF values of the sequenced clones were received by T-RFLP analysis ofcorresponding 16S rRNA and mcrA clones. Noise removal, peak binning to accountfor inter-run differences in T-RF size and normalization of signal intensity wereperformed using R script (R version 2.12.2; http://www.r-project.org) and using acutoff value of six times the standard deviation to remove background noise (17).
RESULTS AND DISCUSSION
Bacterial community composition The bacterial diversity inthe biogas reactor was assessed by constructing 16S rRNA geneclone libraries at two distinct sampling times. 288 clones werecollected in total and screened by restriction fragment lengthpolymorphism analysis. Table 1 illustrates the sequencing results ofrepresentative clones, the next BLAST relatives (to cultured strainsonly), the taxonomic affiliation of the clones based on the RDPClassifier as well as the terminal restriction fragment (T-RF)lengths. Based on the RDP Classifier results and the T-RF values, 9operational taxonomic units (OTUs) were defined. The discoveredbacterial phyla were the Firmicutes comprising 6 OTUs, theBacteroidetes comprising 2 OTUs, and Proteobacteria with 1 OTUdetected. Of the Firmicutes, Clostridia was the most diverse classand comprised members of unknown Clostridia and the generaAnaerotruncus, Tissierella, and Saccharofermentans. Two cloneswere affiliated with unknown Bacteroidetes and one clonebelonged to the phylum Proteobacteria (Castellaniella sp.).
Bacterial community composition was compared with T-RFLPanalysis with two different restriction enzymes, HaeIII and RsaI.Fig. 1 shows the results received after using of HaeIII in T-RFLPanalysis. T-RFs were assigned to OTUs based on the sequence datashown in Table 1. Since one restriction enzyme cannot resolve allOTUs because of the same T-RF lengths of a few phylotypes, for thetaxonomic assignment also the T-RFLP profiles generated with RsaIwere considered (data not shown). T-RFLP allowed the recognitionof about 17e22 different profiles in the reactor. In spite of the factthat some T-RFs could not be taxonomically assigned as they didnot match with any OTUs, most of T-RFs with significant relativeabundances were identified and, therefore, bacteria playing the keyrole in the anaerobic digestion of cattle manure and plant biomasswere established.
The most abundant bacterial phylum detected in the samplesfrom the biogas reactor was Firmicutes, within it Clostridia repre-sented the most diverse group. Within all Clostridia, OTU 3 domi-nated in our reactor and comprised up to 40% of the total T-RF peak
s and experimentally determined terminal restriction fragments (T-RF).
ilarity Taxonomic affiliationaccording to RDP 10
HaeIIIT-RF (bp) RsaIT-RF (bp)
(HM182375)/88% Clostridia 159 e
36903)/91% Clostridia 153 304Clostridiales 234 471Clostridiales 235 471
33)/89% Anaerotruncus sp. 295 10028)/93% Tissierella sp. 297 61
Saccharofermentans sp. 308 7178038)/85% Bacteroidetes 68 3096% Bacteroidetes 255 469
Castellaniella sp. 226 126
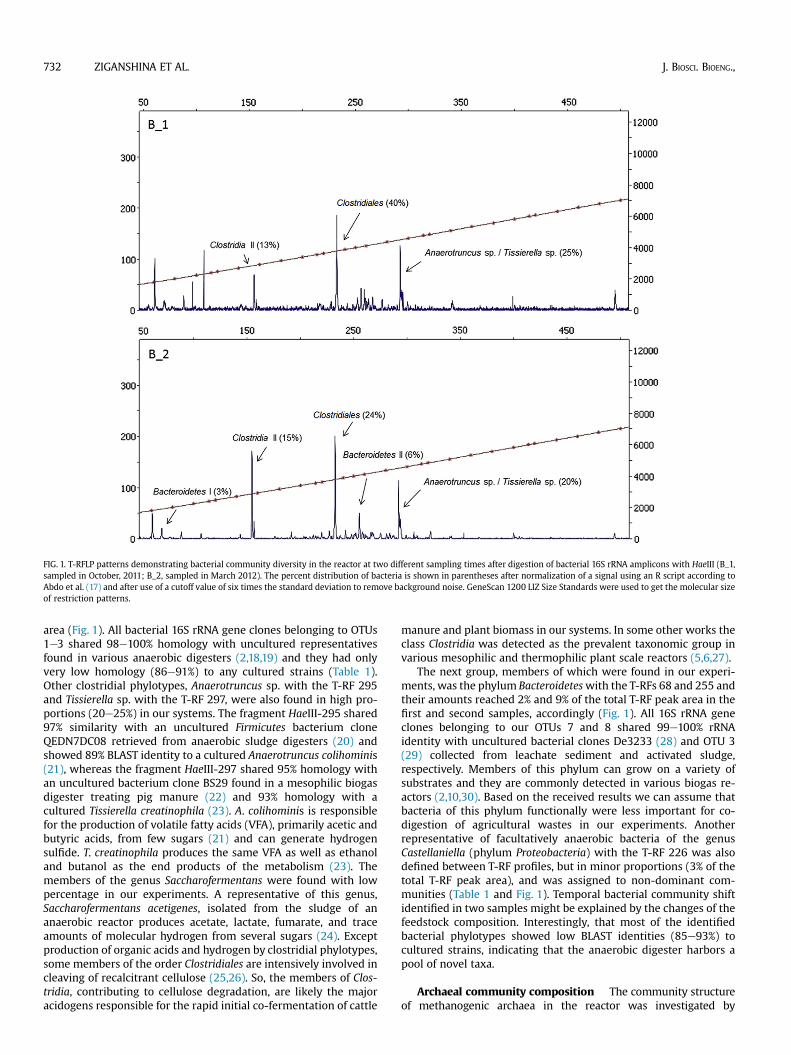
FIG. 1. T-RFLP patterns demonstrating bacterial community diversity in the reactor at two different sampling times after digestion of bacterial 16S rRNA amplicons with HaeIII (B_1,sampled in October, 2011; B_2, sampled in March 2012). The percent distribution of bacteria is shown in parentheses after normalization of a signal using an R script according toAbdo et al. (17) and after use of a cutoff value of six times the standard deviation to remove background noise. GeneScan 1200 LIZ Size Standards were used to get the molecular sizeof restriction patterns.
732 ZIGANSHINA ET AL. J. BIOSCI. BIOENG.,
area (Fig. 1). All bacterial 16S rRNA gene clones belonging to OTUs1e3 shared 98e100% homology with uncultured representativesfound in various anaerobic digesters (2,18,19) and they had onlyvery low homology (86e91%) to any cultured strains (Table 1).Other clostridial phylotypes, Anaerotruncus sp. with the T-RF 295and Tissierella sp. with the T-RF 297, were also found in high pro-portions (20e25%) in our systems. The fragment HaeIII-295 shared97% similarity with an uncultured Firmicutes bacterium cloneQEDN7DC08 retrieved from anaerobic sludge digesters (20) andshowed 89% BLAST identity to a cultured Anaerotruncus colihominis(21), whereas the fragment HaeIII-297 shared 95% homology withan uncultured bacterium clone BS29 found in a mesophilic biogasdigester treating pig manure (22) and 93% homology with acultured Tissierella creatinophila (23). A. colihominis is responsiblefor the production of volatile fatty acids (VFA), primarily acetic andbutyric acids, from few sugars (21) and can generate hydrogensulfide. T. creatinophila produces the same VFA as well as ethanoland butanol as the end products of the metabolism (23). Themembers of the genus Saccharofermentans were found with lowpercentage in our experiments. A representative of this genus,Saccharofermentans acetigenes, isolated from the sludge of ananaerobic reactor produces acetate, lactate, fumarate, and traceamounts of molecular hydrogen from several sugars (24). Exceptproduction of organic acids and hydrogen by clostridial phylotypes,some members of the order Clostridiales are intensively involved incleaving of recalcitrant cellulose (25,26). So, the members of Clos-tridia, contributing to cellulose degradation, are likely the majoracidogens responsible for the rapid initial co-fermentation of cattle
manure and plant biomass in our systems. In some other works theclass Clostridia was detected as the prevalent taxonomic group invarious mesophilic and thermophilic plant scale reactors (5,6,27).
The next group, members of which were found in our experi-ments, was the phylum Bacteroideteswith the T-RFs 68 and 255 andtheir amounts reached 2% and 9% of the total T-RF peak area in thefirst and second samples, accordingly (Fig. 1). All 16S rRNA geneclones belonging to our OTUs 7 and 8 shared 99e100% rRNAidentity with uncultured bacterial clones De3233 (28) and OTU 3(29) collected from leachate sediment and activated sludge,respectively. Members of this phylum can grow on a variety ofsubstrates and they are commonly detected in various biogas re-actors (2,10,30). Based on the received results we can assume thatbacteria of this phylum functionally were less important for co-digestion of agricultural wastes in our experiments. Anotherrepresentative of facultatively anaerobic bacteria of the genusCastellaniella (phylum Proteobacteria) with the T-RF 226 was alsodefined between T-RF profiles, but in minor proportions (3% of thetotal T-RF peak area), and was assigned to non-dominant com-munities (Table 1 and Fig. 1). Temporal bacterial community shiftidentified in two samples might be explained by the changes of thefeedstock composition. Interestingly, that most of the identifiedbacterial phylotypes showed low BLAST identities (85e93%) tocultured strains, indicating that the anaerobic digester harbors apool of novel taxa.
Archaeal community composition The community structureof methanogenic archaea in the reactor was investigated by
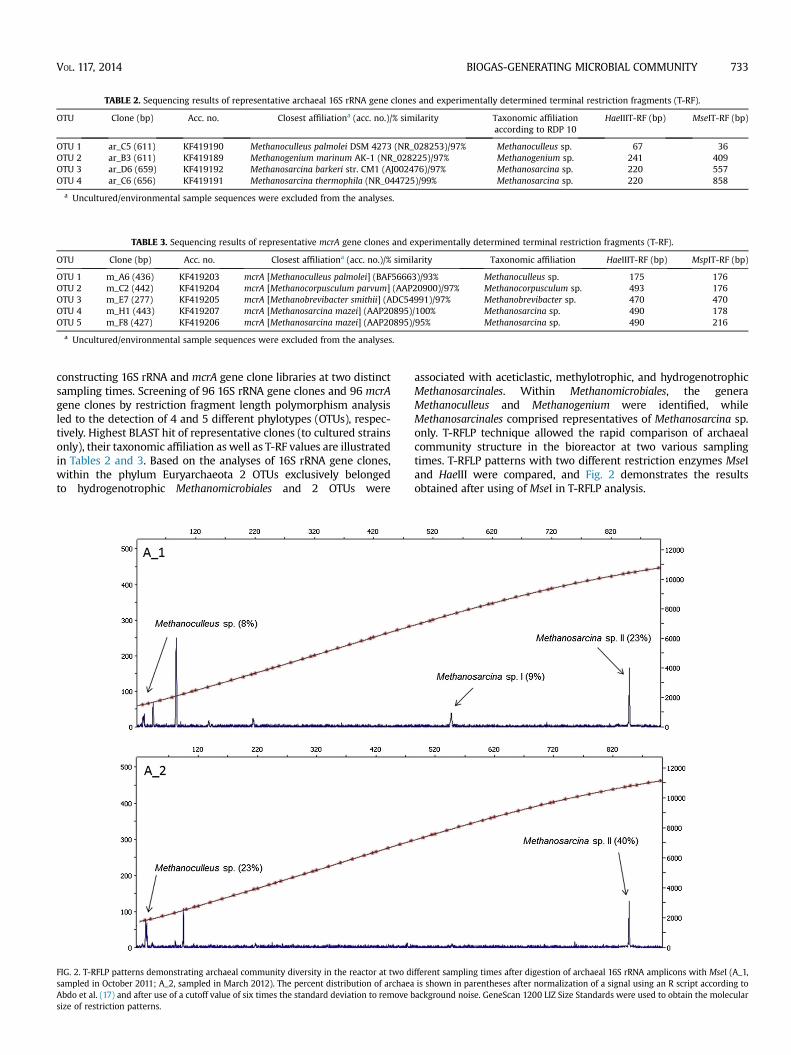
TABLE 2. Sequencing results of representative archaeal 16S rRNA gene clones and experimentally determined terminal restriction fragments (T-RF).
OTU Clone (bp) Acc. no. Closest affiliationa (acc. no.)/% similarity Taxonomic affiliationaccording to RDP 10
HaeIIIT-RF (bp) MseIT-RF (bp)
OTU 1 ar_C5 (611) KF419190 Methanoculleus palmolei DSM 4273 (NR_028253)/97% Methanoculleus sp. 67 36OTU 2 ar_B3 (611) KF419189 Methanogenium marinum AK-1 (NR_028225)/97% Methanogenium sp. 241 409OTU 3 ar_D6 (659) KF419192 Methanosarcina barkeri str. CM1 (AJ002476)/97% Methanosarcina sp. 220 557OTU 4 ar_C6 (656) KF419191 Methanosarcina thermophila (NR_044725)/99% Methanosarcina sp. 220 858
a Uncultured/environmental sample sequences were excluded from the analyses.
TABLE 3. Sequencing results of representative mcrA gene clones and experimentally determined terminal restriction fragments (T-RF).
OTU Clone (bp) Acc. no. Closest affiliationa (acc. no.)/% similarity Taxonomic affiliation HaeIIIT-RF (bp) MspIT-RF (bp)
OTU 1 m_A6 (436) KF419203 mcrA [Methanoculleus palmolei] (BAF56663)/93% Methanoculleus sp. 175 176OTU 2 m_C2 (442) KF419204 mcrA [Methanocorpusculum parvum] (AAP20900)/97% Methanocorpusculum sp. 493 176OTU 3 m_E7 (277) KF419205 mcrA [Methanobrevibacter smithii] (ADC54991)/97% Methanobrevibacter sp. 470 470OTU 4 m_H1 (443) KF419207 mcrA [Methanosarcina mazei] (AAP20895)/100% Methanosarcina sp. 490 178OTU 5 m_F8 (427) KF419206 mcrA [Methanosarcina mazei] (AAP20895)/95% Methanosarcina sp. 490 216
a Uncultured/environmental sample sequences were excluded from the analyses.
VOL. 117, 2014 BIOGAS-GENERATING MICROBIAL COMMUNITY 733
constructing 16S rRNA and mcrA gene clone libraries at two distinctsampling times. Screening of 96 16S rRNA gene clones and 96 mcrAgene clones by restriction fragment length polymorphism analysisled to the detection of 4 and 5 different phylotypes (OTUs), respec-tively. Highest BLAST hit of representative clones (to cultured strainsonly), their taxonomic affiliation as well as T-RF values are illustratedin Tables 2 and 3. Based on the analyses of 16S rRNA gene clones,within the phylum Euryarchaeota 2 OTUs exclusively belongedto hydrogenotrophic Methanomicrobiales and 2 OTUs were
FIG. 2. T-RFLP patterns demonstrating archaeal community diversity in the reactor at two dsampled in October 2011; A_2, sampled in March 2012). The percent distribution of archaeaAbdo et al. (17) and after use of a cutoff value of six times the standard deviation to remove bsize of restriction patterns.
associated with aceticlastic, methylotrophic, and hydrogenotrophicMethanosarcinales. Within Methanomicrobiales, the generaMethanoculleus and Methanogenium were identified, whileMethanosarcinales comprised representatives of Methanosarcina sp.only. T-RFLP technique allowed the rapid comparison of archaealcommunity structure in the bioreactor at two various samplingtimes. T-RFLP patterns with two different restriction enzymes MseIand HaeIII were compared, and Fig. 2 demonstrates the resultsobtained after using of MseI in T-RFLP analysis.
ifferent sampling times after digestion of archaeal 16S rRNA amplicons with MseI (A_1,is shown in parentheses after normalization of a signal using an R script according toackground noise. GeneScan 1200 LIZ Size Standards were used to obtain the molecular
734 ZIGANSHINA ET AL. J. BIOSCI. BIOENG.,
The methanogenic diversity for the mcrA genes was comparedwith the 16S rRNA data. Almost all mcrA sequences as well as 16SrRNA gene sequences received in this work were closely related toclones from anaerobic digesters. In general, a good agreement wasobtained with both approaches, but the rRNA gene method missedthe order Methanobacteriales. According to mcrA genes, 2 OTUsbelonged to Methanomicrobiales (Methanoculleus sp. and Meth-anocorpusculum sp.), 2 OTUs were affiliated withMethanosarcinales(Methanosarcina sp.), and 1 representative was related to hydro-genotrophic Methanobacteriaceae (Methanobrevibacter sp.) in ourexperiments. Fig. 3 shows the patterns generated after digestion ofPCR-amplified products from the total DNA with the restrictionenzyme MspI (results with HaeIII not shown). Since most of themethanogenic T-RFs with high relative abundances were identified,it can be assumed that the key methanogenic archaea involved inanaerobic treating of agricultural organic wastes with biogas pro-duction were identified.
The archaeal community in the reactor was less diverse thanbacterial community and based on the analyses of 16S rRNA genesthe members of the genus Methanosarcina were found at highlevels (up to 40% of the total T-RF peak area in the second sample;Fig. 2). The fragmentsMseI-858 andMseI-557 shared 99% homologywith uncultured archaea clones collected from different digestates(10,31) and compostingmaterials (32). These fragments also shared97e99% homology with cultured Methanosarcina thermophila (33),Methanosarcina barkeri (34), Methanosarcina vacuolata (35),
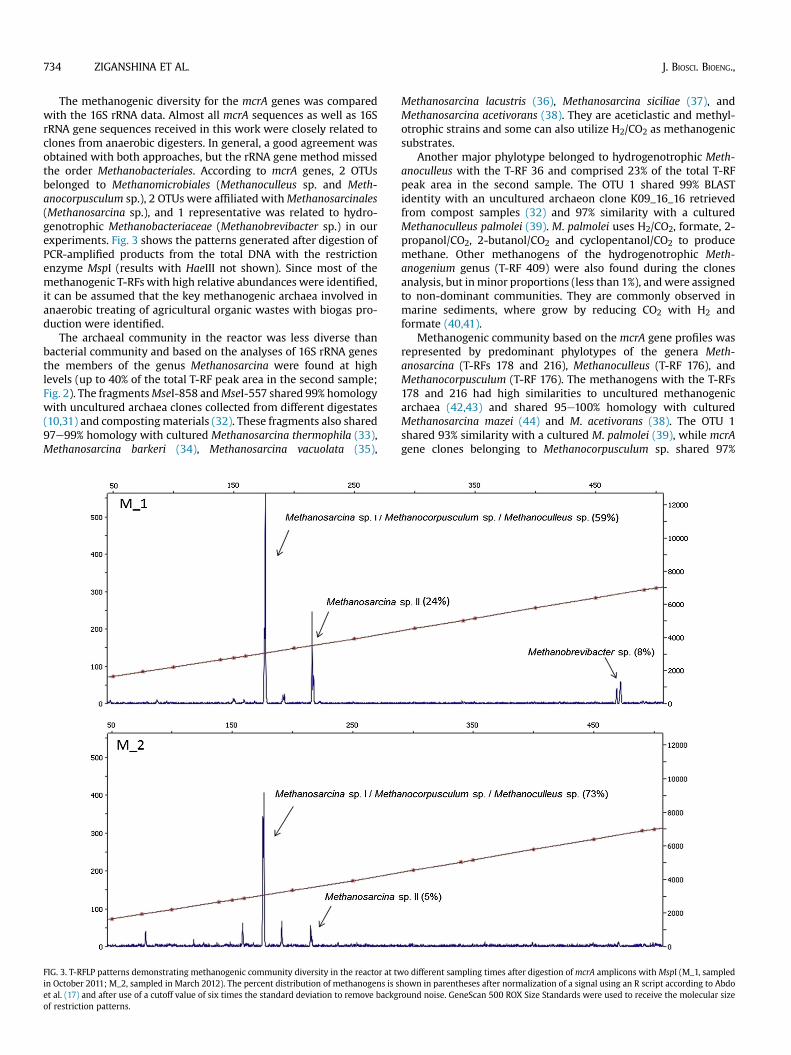
FIG. 3. T-RFLP patterns demonstrating methanogenic community diversity in the reactor at twin October 2011; M_2, sampled in March 2012). The percent distribution of methanogens is set al. (17) and after use of a cutoff value of six times the standard deviation to remove backgof restriction patterns.
Methanosarcina lacustris (36), Methanosarcina siciliae (37), andMethanosarcina acetivorans (38). They are aceticlastic and methyl-otrophic strains and some can also utilize H2/CO2 as methanogenicsubstrates.
Another major phylotype belonged to hydrogenotrophic Meth-anoculleus with the T-RF 36 and comprised 23% of the total T-RFpeak area in the second sample. The OTU 1 shared 99% BLASTidentity with an uncultured archaeon clone K09_16_16 retrievedfrom compost samples (32) and 97% similarity with a culturedMethanoculleus palmolei (39). M. palmolei uses H2/CO2, formate, 2-propanol/CO2, 2-butanol/CO2 and cyclopentanol/CO2 to producemethane. Other methanogens of the hydrogenotrophic Meth-anogenium genus (T-RF 409) were also found during the clonesanalysis, but inminor proportions (less than 1%), andwere assignedto non-dominant communities. They are commonly observed inmarine sediments, where grow by reducing CO2 with H2 andformate (40,41).
Methanogenic community based on the mcrA gene profiles wasrepresented by predominant phylotypes of the genera Meth-anosarcina (T-RFs 178 and 216), Methanoculleus (T-RF 176), andMethanocorpusculum (T-RF 176). The methanogens with the T-RFs178 and 216 had high similarities to uncultured methanogenicarchaea (42,43) and shared 95e100% homology with culturedMethanosarcina mazei (44) and M. acetivorans (38). The OTU 1shared 93% similarity with a cultured M. palmolei (39), while mcrAgene clones belonging to Methanocorpusculum sp. shared 97%
o different sampling times after digestion ofmcrA amplicons with MspI (M_1, sampledhown in parentheses after normalization of a signal using an R script according to Abdoround noise. GeneScan 500 ROX Size Standards were used to receive the molecular size

VOL. 117, 2014 BIOGAS-GENERATING MICROBIAL COMMUNITY 735
BLAST identity with a cultured Methanocorpusculum parvum (45).As can be seen in Figs. 2 and 3, the rRNA gene approach missed theorder Methanobacteriales, whereas analysis of mcrA genes clonesallowed the distinguishing the genus Methanobrevibacter withinthe family Methanobacteriaceae. T-RF 470 had high similarity(100%) to an uncultured archaeon obtained from the rumen ofcattle (46) and 97% sequence homology to Methanobrevibactersmithii, growing and forming methane from either H2/CO2 orformate (47). Methanogenic community shift in our experimentsmight be explained by the changes of the feedstock composition.
Environmental problems associated with the lack of properrecycling of agricultural wastes are some of the major problems inthe world. At the same time these organic wastes are a good po-tential source of bioenergy and can be used as substrates foranaerobic digestion with methane-rich biogas production. Formore stable anaerobic digestion process should be the selection ofwell-adapted microbial populations based on the substratecomposition. With this in mind, modern molecular approachesbased on the detection of species-specific regions within the mi-crobial genome have been applied for characterization of microbialcommunities carrying out the anaerobic digestion (4,8,30).Research results in this field indicate significant progress inimprovement of wastes treatment with biogas production.
The research described herein shows the structure of bacterialand archaeal communities responsible for anaerobic digestion ofthe wastes of agricultural industry, namely cattle manure and plantbiomass. These wastes were subjected to anaerobic microbialconversion in the first pilot-scale biogas reactor in the Republic ofTatarstan. Bacterial and archaeal 16S rRNA genes as well asmethanogen mcrA genes served as markers for the assessment ofmicrobes. Phylogenetic and functional genes diversity was inves-tigated based on the clone libraries and T-RFLP analysis. The resultsof our work clearly show that the cellulose rich feedstock waseffectively digested with some clostridial phylotypes and theywerealso responsible for acidogenesis and acetogenesis stages, whereasmethanogenesis was efficiently performed by the methanogens ofthe genera Methanosarcina and Methanoculleus. The disclosure ofuncultured bacterial phylotypes demonstrating low BLAST identi-ties (to cultivated strains) assumes that the biogas reactor harbors apool of new taxa.
ACKNOWLEDGMENTS
This work was partially supported by the Russian Federal TargetProgram “Research and development on priority directions of sci-entific-technological complex of Russia for 2007e2012” (GC no.16.515.11.5043). Dmitry Belostotskiy’s help with the analyticalprocedures is gratefully acknowledged. Also, the insightfulcomments received during the review process are gratefullyacknowledged.
References
1. Antoni, D., Zverlov, V. V., and Schwarz, W. H.: Biofuels from microbes, Appl.Microbiol. Biotechnol., 77, 23e35 (2007).
2. Kröber, M., Bekel, T., Diaz, N. N., Goesmann, A., Jaenicke, S., Krause, L.,Miller, D., Runte, K. J., Viehöver, P., Pühler, A., and Schlüter, A.: Phylogeneticcharacterization of a biogas plant microbial community integrating clone li-brary 16S-rDNA sequences and metagenome sequence data obtained by 454-pyrosequencing, J. Biotechnol., 142, 38e49 (2009).
3. El-Mashad, H. M. and Zhang, R.: Biogas production from co-digestion of dairymanure and food waste, Bioresour. Technol., 101, 4021e4028 (2010).
4. Karakashev, D., Batstone, D. J., and Angelidaki, I.: Influence of environmentalconditions on methanogenic compositions in anaerobic biogas reactors, Appl.Environ. Microbiol., 71, 331e338 (2005).
5. Goberna, M., Insam, H., and Franke-Whittle, I. H.: Effect of biowaste sludgematuration on the diversity of thermophilic bacteria and archaea in ananaerobic reactor, Appl. Environ. Microbiol., 75, 2566e2572 (2009).
6. Krause, L., Diaz, N. N., Edwards, R. A., Gartemann, K. H., Krömeke, H.,Neuweger, H., Pühler, A., Runte, K. J., Schlüter, A., Stoye, J., and other 3authors: Taxonomic composition and gene content of a methane-producingmicrobial community isolated from a biogas reactor, J. Biotechnol., 136,91e101 (2008).
7. Karlsson, A., Einarsson, P., Schnürer, A., Sundberg, C., Ejlertsson, J., andSvensson, B. H.: Impact of trace element addition on degradation efficiency ofvolatile fatty acids, oleic acid and phenyl acetate and on microbial populationsin a biogas digester, J. Biosci. Bioeng., 114, 446e452 (2012).
8. Wirth, R., Kovács, E., Maróti, G., Bagi, Z., Rákhely, G., and Kovács, K. L.:Characterization of a biogas-producing microbial community by short-readnext generation DNA sequencing, Biotechnol. Biofuels, 5, 1e16 (2012).
9. Qiao, J. T., Qiu, Y. L., Yuan, X. Z., Shi, X. S., Xu, X. H., and Guo, R. B.: Molecularcharacterization of bacterial and archaeal communities in a full-scale anaerobicreactor treating corn straw, Bioresour. Technol., 143, 512e518 (2013).
10. Ziganshin, A. M., Liebetrau, J., Pröter, J., and Kleinsteuber, S.: Microbial com-munity structure and dynamics during anaerobic digestion of various agricul-tural waste materials, Appl. Microbiol. Biotechnol., 97, 5161e5174 (2013).
11. Luton, P. E., Wayne, J. M., Sharp, R. J., and Riley, P. W.: The mcrA gene as analternative to 16S rRNA in the phylogenetic analysis of methanogen pop-ulations in landfill, Microbiology, 148, 3521e3530 (2002).
12. Steinberg, L. M. and Regan, J. M.: Phylogenetic comparison of the methano-genic communities from an acidic, oligotrophic fen and an anaerobic digestertreating municipal wastewater sludge, Appl. Environ. Microbiol., 74,6663e6671 (2008).
13. Hunger, S., Schmidt, O., Hilgarth, M., Horn, M. A., Kolb, S., Conrad, R., andDrake, H. L.: Competing formate- and carbon dioxide-utilizing prokaryotes in ananoxicmethane-emitting fen soil,Appl.Environ.Microbiol.,77, 3773e3785 (2011).
14. Nikolausz, M., Walter, R. F., Sträuber, H., Liebetrau, J., Schmidt, T.,Kleinsteuber, S., Bratfisch, F., Günther, U., and Richnow, H. H.: Evaluation ofstable isotope fingerprinting techniques for the assessment of the predominantmethanogenic pathways in anaerobic digesters, Appl. Microbiol. Biotechnol.,97, 2251e2262 (2013).
15. Shima, S. and Thauer, R. K.: Methyl-coenzyme M reductase and the anaerobicoxidation of methane in methanotrophic Archaea, Curr. Opin. Microbiol., 8,643e648 (2005).
16. Ziganshin, A. M., Schmidt, T., Scholwin, F., Il’inskaya, O. N., Harms, H., andKleinsteuber, S.: Bacteria and archaea involved in anaerobic digestion of dis-tillers grains with solubles, Appl. Microbiol. Biotechnol., 89, 2039e2052 (2011).
17. Abdo, Z., Schüette, U. M., Bent, S. J., Williams, C. J., Forney, L. J., and Joyce, P.:Statistical methods for characterizing diversity of microbial communities byanalysis of terminal restriction fragment length polymorphisms of 16S rRNAgenes, Environ. Microbiol., 8, 929e938 (2006).
18. Tang, Y. Q., Ji, P., Hayashi, J., Koike, Y., Wu, X. L., and Kida, K.: Characteristicmicrobial community of a dry thermophilic methanogenic digester: its long-term stability and change with feeding, Appl. Microbiol. Biotechnol., 91,1447e1461 (2011).
19. Koch, C., Fetzer, I., Schmidt, T., Harms, H., and Muller, S.: Monitoring func-tions in managed microbial systems by cytometric bar coding, Environ. Sci.Technol., 47, 1753e1760 (2013).
20. Riviere, D., Desvignes, V., Pelletier, E., Chaussonnerie, S., Guermazi, S.,Weissenbach, J., Li, T., Camacho, P., and Sghir, A.: Towards the definition of acore of microorganisms involved in anaerobic digestion of sludge, ISME J., 3,700e714 (2009).
21. Lawson, P. A., Song, Y., Liu, C., Molitoris, D. R., Vaisanen, M. L., Collins, M. D.,and Finegold, S. M.: Anaerotruncus colihominis gen. nov., sp. nov., from humanfaeces, Int. J. Syst. Evol. Microbiol., 54, 413e417 (2004).
22. Liu, F. H., Wang, S. B., Zhang, J. S., Zhang, J., Yan, X., Zhou, H. K., Zhao, G. P.,and Zhou, Z. H.: The structure of the bacterial and archaeal community in abiogas digester as revealed by denaturing gradient gel electrophoresis and 16SrDNA sequencing analysis, J. Appl. Microbiol., 106, 952e966 (2009).
23. Harms, C., Schleicher, A., Collins, M. D., and Andreesen, J. R.: Tissierella cre-atinophila sp. nov., a gram-positive, anaerobic, non-spore-forming, creatinine-fermenting organism, Int. J. Syst. Bacteriol., 48, 983e993 (1998).
24. Chen, S., Niu, L., and Zhang, Y.: Saccharofermentans acetigenes gen. nov., sp.nov., an anaerobic bacterium isolated from sludge treating brewery waste-water, Int. J. Syst. Evol. Microbiol., 60, 2735e2738 (2010).
25. Lynd, L. R., Weimer, P. J., van Zyl, W. H., and Pretorius, I. S.: Microbial cel-lulose utilization: fundamentals and biotechnology, Microbiol. Mol. Biol. Rev.,66, 506e577 (2002).
26. Fontes, C. M. and Gilbert, H. J.: Cellulosomes: highly efficient nanomachinesdesigned to deconstruct plant cell wall complex carbohydrates, Annu. Rev.Biochem., 79, 655e681 (2010).
27. Zverlov, V. V., Hiegl, W., Köck, D. E., Kellermann, J., Köllmeier, T., andSchwarz, W. H.: Hydrolytic bacteria in mesophilic and thermophilic degrada-tion of plant biomass, Eng. Life Sci., 10, 528e536 (2010).
28. Liu, J., Wu, W., Chen, C., Sun, F., and Chen, Y.: Prokaryotic diversity, compo-sition structure, and phylogenetic analysis of microbial communities inleachate sediment ecosystems, Appl. Microbiol. Biotechnol., 91, 1659e1675(2011).

736 ZIGANSHINA ET AL. J. BIOSCI. BIOENG.,
29. Li, H., Shen, T. T., Wang, X. L., Lin, K. F., Liu, Y. D., Lu, S. G., Gu, J. D., Wang, P.,Lu, Q., and Du, X. M.: Biodegradation of perchloroethylene and chlorophenolco-contamination and toxic effect on activated sludge performance, Bioresour.Technol., 137, 286e293 (2013).
30. Li, A., Chu, Y., Wang, X., Ren, L., Yu, J., Liu, X., Yan, J., Zhang, L., Wu, S., andLi, S.: A pyrosequencing-based metagenomic study of methane-producingmicrobial community in solid-state biogas reactor, Biotechnol. Biofuels, 6,1e17 (2013).
31. Nunoura, T., Oida, H., Miyazaki, J., Miyashita, A., Imachi, H., and Takai, K.:Quantification of mcrA by fluorescent PCR in methanogenic and methano-trophic microbial communities, FEMS Microbiol. Ecol., 64, 240e247 (2008).
32. Yamamoto, N., Asano, R., Yoshii, H., Otawa, K., and Nakai, Y.: Archaealcommunity dynamics and detection of ammonia-oxidizing archaea duringcomposting of cattle manure using culture-independent DNA analysis, Appl.Microbiol. Biotechnol., 90, 1501e1510 (2011).
33. Zinder, S. H., Sowers, K. R., and Ferry, J. G.: Methanosarcina thermophila sp.nov., a thermophilic, acetotrophic, methane-producing bacterium, Int. J. Syst.Bacteriol., 35, 522e523 (1985).
34. Maestrojuan, G. M. and Boone, D. R.: Characterization of Methanosarcinabarkeri MST and 227, Methanosarcina mazei S-6T, and Methanosarcina vacuolataZ-761T, Int. J. Syst. Bacteriol., 41, 267e274 (1991).
35. Zhilina, T. N. and Zavarzin, G. A.: Methanosarcina vacuolata sp. nov. a vacuo-lated Methanosarcina, Int. J. Syst. Bacteriol., 37, 281e283 (1987).
36. Simankova, M. V., Parshina, S. N., Tourova, T. P., Kolganova, T. V.,Zehnder, A. J., and Nozhevnikova, A. N.: Methanosarcina lacustris sp. nov., anew psychrotolerant methanogenic archaeon from anoxic lake sediments, Syst.Appl. Microbiol., 24, 362e367 (2001).
37. Elberson, M. A. and Sowers, K. R.: Isolation of an aceticlastic strain of Meth-anosarcina siciliae from marine canyon sediments and emendation of thespecies description for Methanosarcina siciliae, Int. J. Syst. Bacteriol., 47,1258e1261 (1997).
38. Sowers, K. R., Baron, S. F., and Ferry, J. G.: Methanosarcina acetivorans sp. nov.,an acetotrophic methane-producing bacterium isolated from marine sedi-ments, Appl. Environ. Microbiol., 47, 971e978 (1984).
39. Zellner, G., Messner, P., Winter, J., and Stackebrandt, E.: Methanoculleuspalmolei sp. nov., an irregularly coccoid methanogen from an anaerobicdigester treating wastewater of a palm oil plant in north-Sumatra, Indonesia,Int. J. Syst. Bacteriol., 48, 1111e1117 (1998).
40. Romesser, J. A., Wolfe, R. S., Mayer, F., Spiess, E., and Walther-Mauruschat, A.: Methanogenium, a new genus of marine methanogenic bac-teria, and characterization of Methanogenium cariaci sp. nov. and Meth-anogenium marisnigri sp. nov., Arch. Microbiol., 121, 147e153 (1979).
41. Kendall, M. M., Wardlaw, G. D., Tang, C. F., Bonin, A. S., Liu, Y., andValentine, D. L.: Diversity of archaea in marine sediments from Skan Bay,Alaska, including cultivated methanogens, and description of Methanogeniumboonei sp. nov., Appl. Environ. Microbiol., 73, 407e414 (2007).
42. Roussel, E. G., Sauvadet, A. L., Allard, J., Chaduteau, C., Richard, P., CambonBonavita, M. A., and Chaumillon, E.: Archaeal methane cycling communitiesassociated with gassy subsurface sediments of Marennes-Oléron bay (France),Geomicrobiol. J., 26, 31e43 (2009).
43. Newberry, C. J., Webster, G., Cragg, B. A., Parkes, R. J., Weightman, A. J., andFry, J. C.: Diversity of prokaryotes and methanogenesis in deep subsurfacesediments from the Nankai Trough, Ocean Drilling Program Leg 190, Environ.Microbiol., 6, 274e287 (2004).
44. Blotevogel, K. H. and Fischer, U.: Transfer of Methanococcus frisius to thegenusMethanosarcina asMethanosarcina frisia comb. nov., Int. J. Syst. Bacteriol.,39, 91e92 (1989).
45. Zellner, G., Alten, C., Stackebrandt, E., Conway de Macario, E., and Winter, J.:Isolation and characterization of Methanocorpusculum parvum, gen. nov., spec.nov., a new tungsten requiring, coccoid methanogens, Arch. Microbiol., 147,13e20 (1987).
46. Denman, S. E., Tomkins, N. W., and McSweeney, C. S.: Quantitation and di-versity analysis of ruminal methanogenic populations in response to theantimethanogenic compound bromochloromethane, FEMS Microbiol. Ecol., 62,313e322 (2007).
47. Miller, T. L., Wolin, M. J., de Macario, E. C., and Macario, A. J. L.: Isolation ofMethanobrevibacter smithii from human feces, Appl. Environ. Microbiol., 43,227e232 (1982).