apoptosis - krooart.com fileapoptosis! disrupted vs. intact cell membrane in necrosis and apoptosis...
TRANSCRIPT
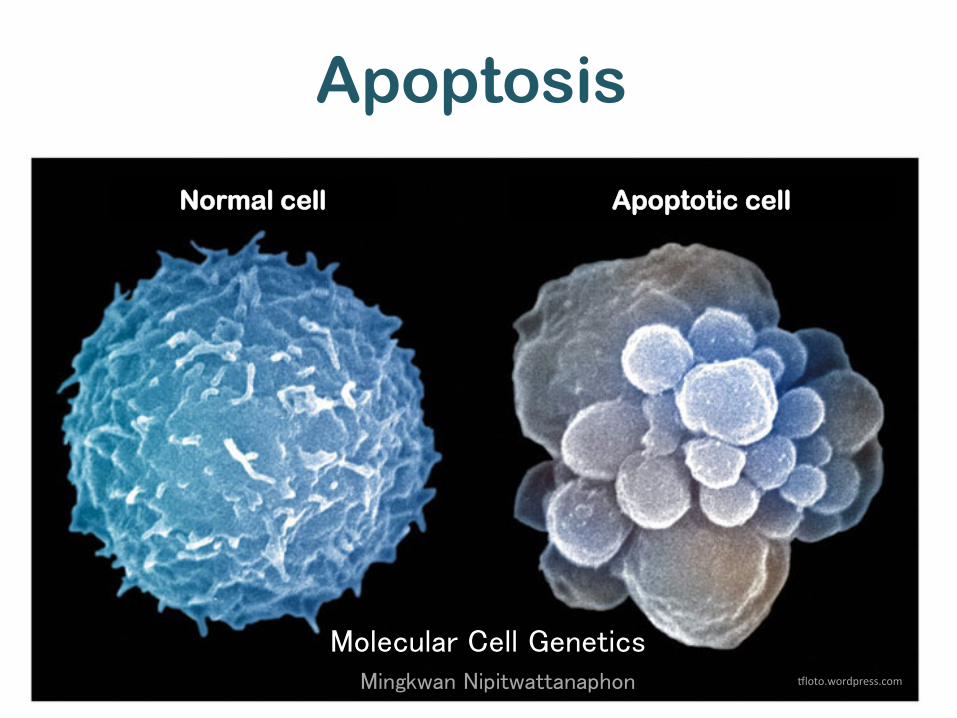
Apoptosis
Molecular Cell Genetics
Mingkwan Nipitwattanaphon "loto.wordpress.com
Normal cell Apoptotic cell

Programmed cell-death (or PCD)
• Death of cells mediated by intracellular program • 3 main types of PCD
Apoptosis – the best-characterized type of PCD because of its importance in
development and homeostasis Autophagy – selective degradation of intracellular targets, such as misfolded proteins
and damaged organelles, and is an important homeostatic function. Necrosis – an uncontrolled cell death characterized by cell swelling, as well as
destruction of the plasma membrane and subcellular organelles – An alternate form of PCD caused by external factors such as
pathogens, lack of oxygen, or blood supply.

Necrosis • Caused by
– Damaged plasma membrane – Lack of oxygen supply – Insufficient ATP synthesis
• CharacterisBcs – Swelling – formaBon of cytoplasmic vacuoles – distended endoplasmic reBculum – formaBon of cytoplasmic blebs – Condensed – swollen or ruptured mitochondria – disaggregaBon and detachment of ribosomes – disrupted organelle membranes – swollen and ruptured lysosomes – disrupBon of the cell membrane

Apoptotic cells
• Lose contact to neighbor cells and shrink • protein cleavage • Protein cross-‐linking • DNA breakdown (∼180-‐200bp) by Ca2+ and Mg2+-‐dependent endonucleases
• Presence of “eat me” signal (phosphaBdylserine), Annexin I, calreBculin, on their surface
• phagocyBc recogniBon

Apoptotic characteristics
• Cell shrinkage -‐ the cytoplasm is dense and the organelles are more Bghtly packed
• Pyknosis = irreversible chromaBn condensaBon (found in both necrosis and apoptosis)
• Histological examinaBon: hematoxylin and eosin stain – dark eosinophilic cytoplasm and dense purple nuclear chromaBn fragments
• Extensive plasma membrane blebbing (irregular bulge in the plasma membrane of a cell, caused by localized decoupling of the cytoskeleton from the plasma membrane)
• karyorrhexis (chromaBn fragmentaBon) • separaBon of cell fragments into apoptoBc bodies (budding).

Apoptosis vs. Necrosis
• Although different, very similar, have a lot of overlapping processes, e.g. DNA fragmentaBon – Apoptosis – cell shrinkage and suicide, no damage to other cells, high level of transglutaminase acBvity (cross-‐linking proteins)
– Necrosis – cell swells and bursts -‐> inflammatory response. • Whether cells are going to apoptosis or necrosis someBmes
depend on the type of and/or degree of sBmuli • Apoptosis is geneBcally determined • apoptosis is energy-‐dependent process that involves the
acBvaBon of a group of cysteine proteases called “caspases”

Apoptosis vs. Necrosis
h]p://en.wikipedia.org/wiki/Necrosis
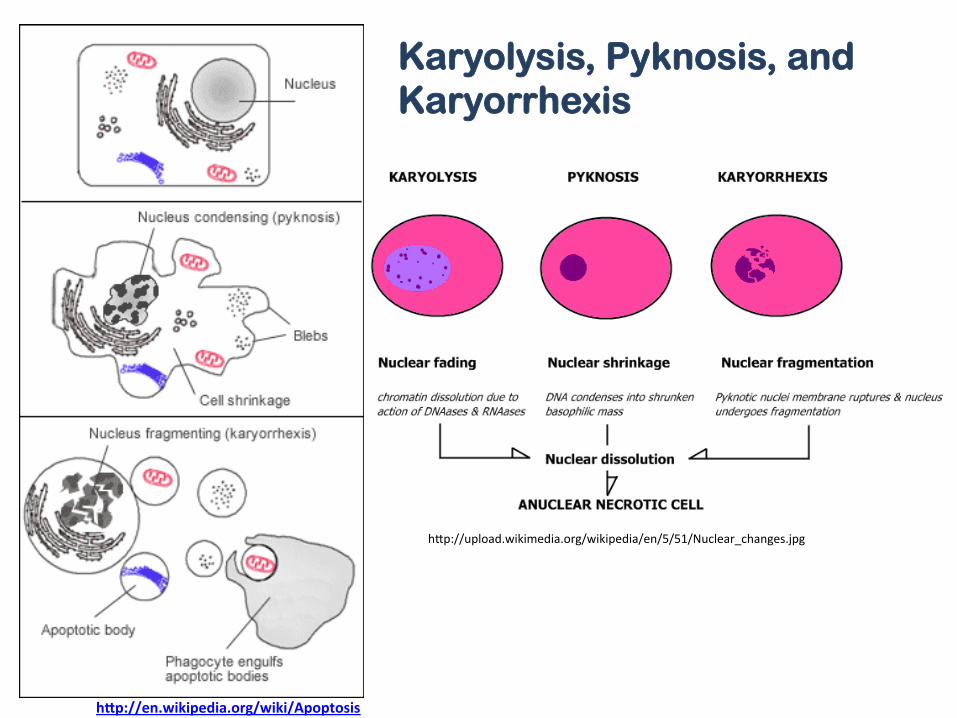
h]p://upload.wikimedia.org/wikipedia/en/5/51/Nuclear_changes.jpg
h"p://en.wikipedia.org/wiki/Apoptosis
Karyolysis, Pyknosis, and Karyorrhexis

Apoptosis vs. Necrosis
Energy-dependent mode of death
Energy-independent
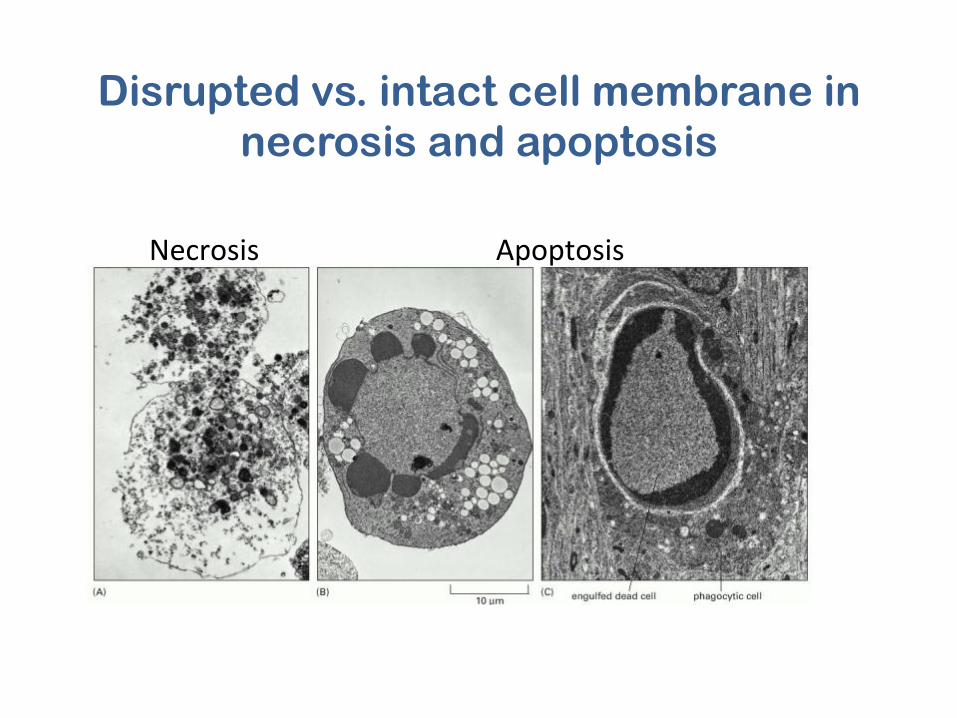
Necrosis Apoptosis
Disrupted vs. intact cell membrane in necrosis and apoptosis

Dark eosinophilic cytoplasm and dense purple nuclear chromatin fragments in apoptotic cells
Elmore S. Toxicologic Pathology, 35:495–516, 2007

Other types of PCDs
• Aponecrosis (necropoptosis) – shared dynamic, molecular, and morphological features with both
apoptosis and necrosis – found when the cells were exposed to high doses, of the hypoxic
stimulus – an incomplete execution of the apoptotic program and the following
degeneration in necrosis. • Paraptosis
– morphologically distinct from apoptosis and necrosis – cytoplasmic vacuolation and late mitochondrial swelling and clumping
(size of vacuole increases over time) – independent of caspase activation and inhibition – lack of apoptotic morphology, e.g. membrane blebbing, chromatin
condensation, and nuclear fragmentation
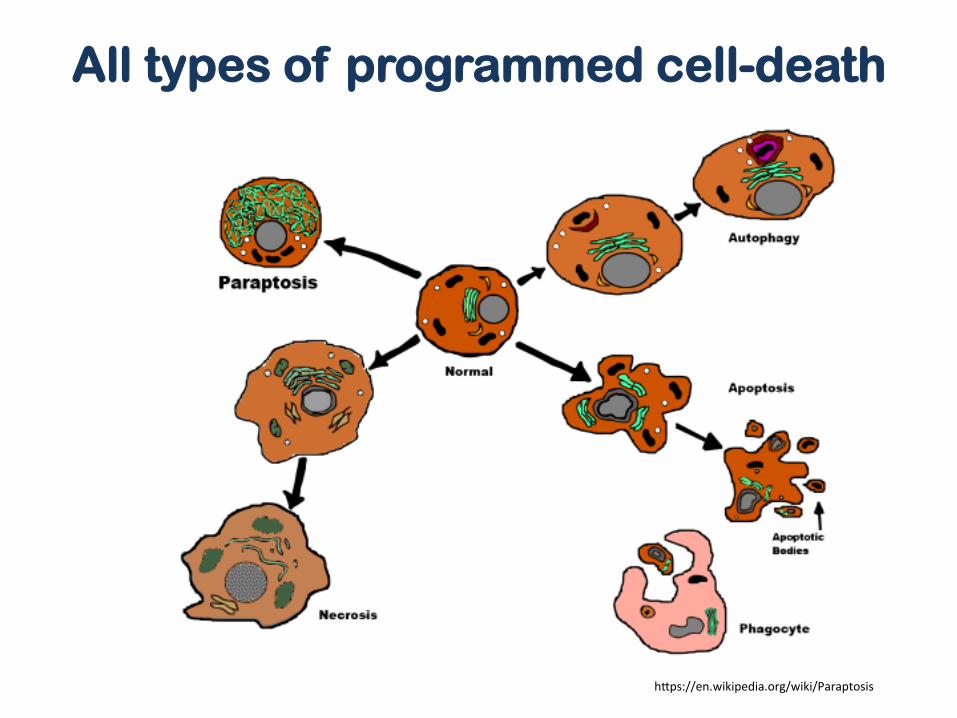
h]ps://en.wikipedia.org/wiki/Paraptosis
All types of programmed cell-death

Other types of PCDs
• Pyronecrosis – Similar to necrosis but some different factors
are involved – Not involved in innate immune system
• Entosis – a form of ‘cellular cannibalism’ in patient who
has Huntington's disease


Importance of Apoptosis
• normal cell turnover • proper development • functioning of the immune system • hormone-dependent atrophy • embryonic development and chemical-
induced cell death. • Inappropriate apoptosis (either too little or too
much) will lead to neurodegenerative diseases, ischemic damage, autoimmune disorders and many types of cancer.

Roles of apoptosis
• sculpting structures • deleting structures • regulating cell number • eliminating defective cells
suggest that autophagy also contributes to the removal of super-fluous cells during normal development (reviewed in Ryoo andBaehrecke, 2010).
As one may expect, the regulation of PCD in vertebratesappears considerably more complex, and vast numbers of cellsundergo PCD throughout development, from as early as innercell mass differentiation in blastocysts to maintenance of tissuehomeostasis in adulthood (Hardy et al., 1989). Therefore, it issomewhat surprising that the inactivation of mouse cell deathgenes leads to only relatively minor developmental defectsand can often survive embryonic development (see, for ex-ample, Lindsten and Thompson, 2006; Okamoto et al., 2006).One reason appears to be considerable redundancy within thecaspase family and the existence of multiple mechanisms forcaspase activation. For example, some effector caspases canbe activated in the absence of Apaf-1 function (Nagasakaet al., 2010). In addition, there is evidence for alternative backupmechanisms that eliminate cells when apoptosis is defective(reviewed in Yuan and Kroemer, 2010). Despite the apparentrobustness of cell death mechanisms in mammals, inhibitionof apoptosis has been linked to several specific develop-mental abnormalities and also a variety of human pathol-ogies, including cancer and degenerative diseases (Thompson,1995).
Studies in worms, flies, and mice have been complementedand extended by work in many other systems, including Hydra,Manduca, Xenopus, zebrafish, chicken, and the analysis ofhuman patients. Collectively, this work has illustrated why cellsneed to be eliminated in different physiological contexts: (1)sculpting and (2) deleting structures, (3) regulating cell number,and (4) eliminating defective cells.
Sculpting Structures and Driving MorphogenesisPCDplays a crucial role in organogenesis and tissue remodeling.Perhaps the best-known example is the formation of digitsin higher vertebrates in which PCD eliminates the interdigitalwebs primarily via the apoptotic machinery (Figure 2A; Lindstenet al., 2000). Although apoptosis is the major cell death mecha-nism in developing limbs, inactivation of proapoptotic genes inthe mouse only partially prevents the removal of the interdigitaltissue, suggesting that backup mechanisms exist when apo-ptosis fails (Yuan and Kroemer, 2010). In Drosophila, apoptosisplays a critical role in the formation of leg joints and for themorphogenesis of segments, in particular of the head, whichall require RHG-mediated apoptosis (Lohmann et al., 2002).Furthermore, apoptosis is also required to permit tissue rotationthat drives looping morphogenesis of male genitalia in the fly(Kuranaga et al., 2011; Suzanne et al., 2010).
PCD is also involved in the conversion of solid structures tohollow tubes, thereby yielding lumina such as in the creation ofthe proamniotic cavity (Coucouvanis and Martin, 1995; Weilet al., 1997). It is observed when epithelial sheets invaginate,forming tubes or vesicles, for example, in the establishment ofthe neural tube or lens and when epithelial sheets fuse to con-struct the mammalian palate (Glucksmann, 1951). In addition,PCD is involved in sculpting the future inner ear in chicks (Aval-lone et al., 2002) and is essential for generating the four-chamberarchitecture of the heart (Abdelwahid et al., 2002).
Deleting StructuresDuring development, various structures that serve a transientfunction are removed by PCD when they are no longer required.Examples include evolutionary relics, structures that are re-quired in only one sex, or structures that are transiently required.In fish and amphibians, pronerphric tubules form functioningkidneys; however, they are not utilized in mammals and arehence eliminated during embryogenesis. In female mammals,the Mullerian duct forms the oviducts and uterus but is deletedin males. Conversely, the Wolffian duct that forms the vas
Figure 2. Functions of PCD during Development(A) PCD regulates proper structure sculpting by eliminating interdigitalwebbings.(B) During Drosophila metamorphosis, nearly all larval structures are de-stroyed, such as the salivary glands (SG), muscles (M), midgut (MG), andhindgut (HG) (depicted by purple), whereas novel structures are raised fromundifferentiated cells termed imaginal discs (depicted by various colors).The locations and developmental fates of the imaginal discs are similarlyillustrated.(C and D) PCD also controls cell number, for example, by deleting cells that failto partner (C) and eliminates dangerous and abnormal cells such as autor-eactive lymphocytes (D).
744 Cell 147, November 11, 2011 ª2011 Elsevier Inc.
Fuchs & Steller (2011) Cell 147: 742-758

1. sculpting structures
• conversion of solid structures to hollow tubes, e.g.: – proamniotic cavity – establishment of the neural tube or lens – epithelial sheets fuse to construct the
mammalian palate • sculpting the future inner ear in chicks • generating the four-chamber architecture
of the heart

• conversion of solid structures to hollow tubes, e.g. proamnioBc cavity, establishment of the neural tube or lens and when epithelial sheets fuse to construct the mammalian palate
• sculpBng the future inner ear in chicks • generaBng the four-‐chamber architecture of the heart
1. sculpting structures: lens

1. sculpting structures: proamniotic cavity
From sqqd.dromibd.top

SchemaBc model of the process of cavitaBon based on the formaBon of the pro-‐amnioBc cavity in mouse embryos
ProamnioBc cavity
1. sculpting structures: proamniotic cavity
H E Abud Cell Death and DifferenBaBon (2004) 11, 797–799

h]p://www.bionalogy.com/eye_and_ear_files/image003.jpg
1. sculpting structures: inner ear development

1. sculpting structures: heart chambers

h]p://www.ncbi.nlm.nih.gov/books/NBK26873/
1. Sculpting structures: fingers

h]p://www.ncbi.nlm.nih.gov/books/NBK26873/
2. deleting structures: metamorphosis

h]p://www.ncbi.nlm.nih.gov/books/NBK26873/
2. deleting structures: metamorphosis
suggest that autophagy also contributes to the removal of super-fluous cells during normal development (reviewed in Ryoo andBaehrecke, 2010).
As one may expect, the regulation of PCD in vertebratesappears considerably more complex, and vast numbers of cellsundergo PCD throughout development, from as early as innercell mass differentiation in blastocysts to maintenance of tissuehomeostasis in adulthood (Hardy et al., 1989). Therefore, it issomewhat surprising that the inactivation of mouse cell deathgenes leads to only relatively minor developmental defectsand can often survive embryonic development (see, for ex-ample, Lindsten and Thompson, 2006; Okamoto et al., 2006).One reason appears to be considerable redundancy within thecaspase family and the existence of multiple mechanisms forcaspase activation. For example, some effector caspases canbe activated in the absence of Apaf-1 function (Nagasakaet al., 2010). In addition, there is evidence for alternative backupmechanisms that eliminate cells when apoptosis is defective(reviewed in Yuan and Kroemer, 2010). Despite the apparentrobustness of cell death mechanisms in mammals, inhibitionof apoptosis has been linked to several specific develop-mental abnormalities and also a variety of human pathol-ogies, including cancer and degenerative diseases (Thompson,1995).
Studies in worms, flies, and mice have been complementedand extended by work in many other systems, including Hydra,Manduca, Xenopus, zebrafish, chicken, and the analysis ofhuman patients. Collectively, this work has illustrated why cellsneed to be eliminated in different physiological contexts: (1)sculpting and (2) deleting structures, (3) regulating cell number,and (4) eliminating defective cells.
Sculpting Structures and Driving MorphogenesisPCDplays a crucial role in organogenesis and tissue remodeling.Perhaps the best-known example is the formation of digitsin higher vertebrates in which PCD eliminates the interdigitalwebs primarily via the apoptotic machinery (Figure 2A; Lindstenet al., 2000). Although apoptosis is the major cell death mecha-nism in developing limbs, inactivation of proapoptotic genes inthe mouse only partially prevents the removal of the interdigitaltissue, suggesting that backup mechanisms exist when apo-ptosis fails (Yuan and Kroemer, 2010). In Drosophila, apoptosisplays a critical role in the formation of leg joints and for themorphogenesis of segments, in particular of the head, whichall require RHG-mediated apoptosis (Lohmann et al., 2002).Furthermore, apoptosis is also required to permit tissue rotationthat drives looping morphogenesis of male genitalia in the fly(Kuranaga et al., 2011; Suzanne et al., 2010).
PCD is also involved in the conversion of solid structures tohollow tubes, thereby yielding lumina such as in the creation ofthe proamniotic cavity (Coucouvanis and Martin, 1995; Weilet al., 1997). It is observed when epithelial sheets invaginate,forming tubes or vesicles, for example, in the establishment ofthe neural tube or lens and when epithelial sheets fuse to con-struct the mammalian palate (Glucksmann, 1951). In addition,PCD is involved in sculpting the future inner ear in chicks (Aval-lone et al., 2002) and is essential for generating the four-chamberarchitecture of the heart (Abdelwahid et al., 2002).
Deleting StructuresDuring development, various structures that serve a transientfunction are removed by PCD when they are no longer required.Examples include evolutionary relics, structures that are re-quired in only one sex, or structures that are transiently required.In fish and amphibians, pronerphric tubules form functioningkidneys; however, they are not utilized in mammals and arehence eliminated during embryogenesis. In female mammals,the Mullerian duct forms the oviducts and uterus but is deletedin males. Conversely, the Wolffian duct that forms the vas
Figure 2. Functions of PCD during Development(A) PCD regulates proper structure sculpting by eliminating interdigitalwebbings.(B) During Drosophila metamorphosis, nearly all larval structures are de-stroyed, such as the salivary glands (SG), muscles (M), midgut (MG), andhindgut (HG) (depicted by purple), whereas novel structures are raised fromundifferentiated cells termed imaginal discs (depicted by various colors).The locations and developmental fates of the imaginal discs are similarlyillustrated.(C and D) PCD also controls cell number, for example, by deleting cells that failto partner (C) and eliminates dangerous and abnormal cells such as autor-eactive lymphocytes (D).
744 Cell 147, November 11, 2011 ª2011 Elsevier Inc.

h]p://scienceblogs.com/pharyngula/2011/05/02/the-‐basics-‐of-‐building-‐a-‐kidne/
• pronerphric tubules of kidneys are eliminated during embryogenesis • In female mammals, the Müllerian duct forms the oviducts and uterus but is
deleted in males. • In contrast, Wolffian duct that forms the vasdeferens, epididymis, and seminal
vesicle in males is degraded in females
2. deleting structures: kidney, sex organs Degenerating mesonephros (Woffian duct)
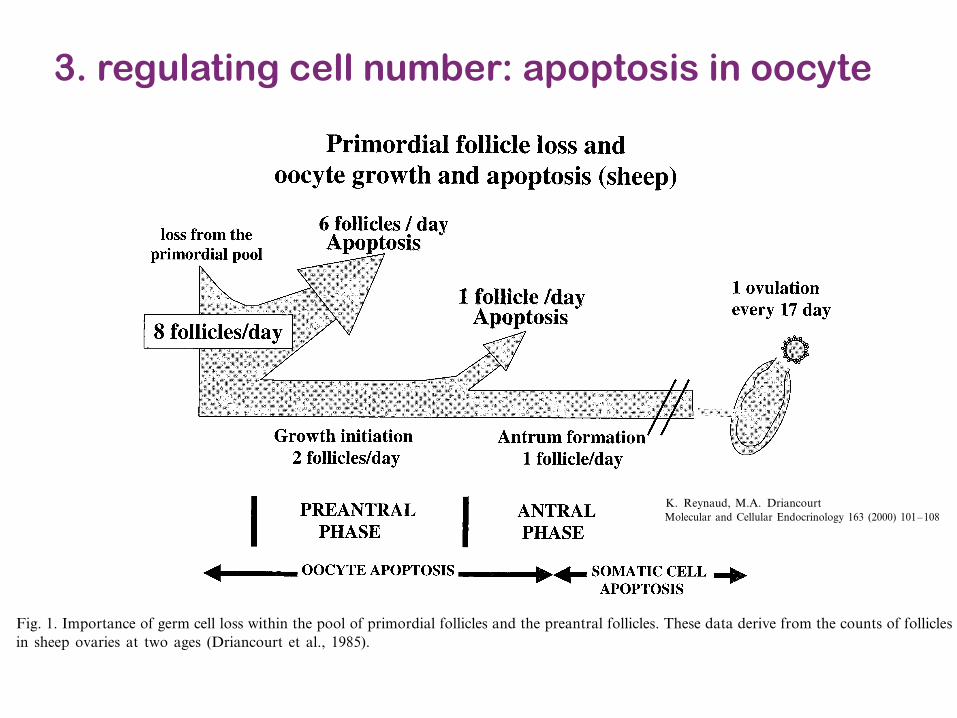
3. regulating cell number: apoptosis in oocyte
K. Reynaud, M.A. Driancourt / Molecular and Cellular Endocrinology 163 (2000) 101–108102
2. Cell death in the ovary
2.1. During oogenesis
In all species, peak numbers of germ cells are ob-served around the time of the mitotic to meiotic transi-tion (Gondos, 1978). At this stage, the mean germ cellstore of a mouse, cow or woman ovary is 2.5×105,2.1×106 and 6.8×106 germ cells, respectively (Baker,1963; Erickson, 1966; Tam and Snow, 1981). In vivoresults (i.e. visualization of ‘atretic divisions’, Baker,1963) suggest that the rapid proliferation of oogoniamay be associated with germ cell loss. Furthermore,strong evidence for germ cell loss amongst the popula-tion of proliferative oogonia has been obtained in vitro(Coucouvanis et al., 1993; Pesce et al., 1993). Distinc-tive features of the degenerating cells are condensednuclei with clumps of dense chromatin located at thenuclear periphery. From their peak number observed atE13, E80–110 and fifth month of pregnancy (mouse,cattle and women, respectively), the number of germcells sharply decreases with two main periods of highgerm cell loss, the pachytene stage of meiosis in oocytesand the formation of the primordial follicles (Gondos,1978). As a consequence, the number of germ cellsenclosed in primordial follicles at birth is less than 20%(human, Baker, 1963) or less than 5% (cow, Erickson,1966) of its peak number. This is solid evidence demon-strating that the normal fate for a female germ cellduring oogenesis is death.
2.2. During folliculogenesis
In all species, the store of primordial follicles de-creases with time. In women, menopause is the conse-quence of the exhaustion of the pool of such follicles.Gougeon et al. (1994), using mathematical modeling ofthe decrease in the size of the primordial pool withtime, have demonstrated that in women younger than38-years-old, this decrease is produced by growth initia-tion and oocyte loss while, in older women, it is solelygenerated by growth initiation occurring at a fasterpace.
In sheep, comparison of the population of primordialfollicles in ovaries of 2 and 8-year-old ewes has demon-strated that eight primordial follicles disappear fromthe reserve pool every day (Driancourt et al., 1985).This contrasts with the estimated rate of initiation ofgrowth from the pool (two and three follicles per day)and strongly suggests that five to six follicles die everyday within the primordial pool. Since granulosa cellapoptosis is never visualized in such follicles, it may beassumed that oocyte death is the cause of this highdeath rate within primordial follicles (Fig. 1). A similarreasoning demonstrates that 50% of the preantral folli-cles disappear through the preantral stage (Driancourtet al., 1985; Fig. 1).
Although these mathematical considerations stronglysupport the idea that oocyte death occurs within pri-mordial and preantral follicles until recently, there werefew data firmly demonstrating it. This is mainly becausepycnotic bodies, the most common marker used to
Fig. 1. Importance of germ cell loss within the pool of primordial follicles and the preantral follicles. These data derive from the counts of folliclesin sheep ovaries at two ages (Driancourt et al., 1985).
Molecular and Cellular Endocrinology 163 (2000) 101–108
Oocyte attrition
K. Reynaud, M.A. Driancourt *INRA-URA CNRS 1291, PRMD, 37380 Nouzilly, France
Received 25 June 1999; received in revised form 6 August 1999; accepted 29 September 1999
Abstract
During oogenesis, germ cell numbers sharply decrease when meiosis is initiated. There is solid evidence (DNA ladders, in situdetection) that this loss is through apoptosis. Oocyte apoptosis appears to hit mitotic primordial germ cells (PGC), pachyteneoocytes and early primordial follicles. The control of oocyte apoptosis is not fully understood, although survival factors (LIF, kitligand and FGF), as well as death inducing factors (fas ligand, TGF!), have been identified. Fas ligand binding on oocytic fasmay result in caspase 8 activation. Two pathways inducing oocyte apoptosis may then be operating. In the first one, activatedcaspase 8 will induce activation of executioner caspases. In the second one, activated caspase 8 will trigger the cleavage of the bcl2family member Bid, which will act on mitochondria, resulting in cytochrome c release, caspase 9 activation and finally, activationof all executioner caspases. As a consequence of caspase activation, alterations in the cell nucleus (DNAse activation, PARPfragmentation), in the cell cytoskeleton (lamin) and cell metabolism will occur, producing cell death. During folliculogenesis, germcell loss, owing to oocyte apoptosis, has been postulated within primordial and preantral follicles. Its regulatory mechanisms maybe even more complex than those operating in foetal oocytes since additional control factors include EGF/TGF" and bcl2(survival) and activin (death inducer). In contrast, oocytes from antral follicles appear to be very unsensitive to death inducingstimuli. © 2000 Elsevier Science Ireland Ltd. All rights reserved.
Keywords: Oocyte; Apoptosis; Follicle
www.elsevier.com/locate/mce
1. Introduction
Females of all domestic species and primates have afinite store of germ cells, which is established duringoogenesis and used later for folliculogenesis. Formationof this store (i.e. the pool of primordial follicles) gener-ally occurs during foetal life as a result of a number ofoverlapping events, migration of the primordial germcells (PGC) to the gonadal ridge, proliferation of thesePGC, initiation of meiotic prophase followed by itsblock to the diplotene stage, and finally, formation ofthe primordial follicles, when cells originating from therete ovarii surround the oocytes. Germ cell loss appearshowever to occur at each of these steps. The first
section of this review will attempt to clarify how andwhy germ cell loss occurs throughout oogenesis.
Ewes and women ovulate every 17 and 28 days,respectively. This infrequent occurrence of ovulationcontrasts with the continuous exit from the reserve ofprimordial follicles (2 and 10–30 follicles initiategrowth) every day in sheep and in young women,respectively, according to Driancourt et al. (1985),Faddy and Gosden (1995). This suggests that a verystrong tuning of the number of growing follicles occursthroughout folliculogenesis. The respective contributionof oocyte apoptosis versus apoptosis of somatic cells togenerate a single ovulatory follicle from a continuousflux of growing follicles will be detailed in the secondsection of this review. For clarity, only three specieswhich differ in the timings of their phases of oogenesis(mouse versus sheep and humans) or in their strategiesto use their pool of primordial follicles for folliculogen-esis (mouse and sheep versus humans who displaymenopause) will be considered.
* Corresponding author. Tel.: +33-2-41228265; fax: +33-2-41228251.
E-mail address: [email protected](M.A. Driancourt).
0303-7207/00/$ - see front matter © 2000 Elsevier Science Ireland Ltd. All rights reserved.PII: S0303 -7207 (99 )002 46 -4
Molecular and Cellular Endocrinology 163 (2000) 101–108
Oocyte attrition
K. Reynaud, M.A. Driancourt *INRA-URA CNRS 1291, PRMD, 37380 Nouzilly, France
Received 25 June 1999; received in revised form 6 August 1999; accepted 29 September 1999
Abstract
During oogenesis, germ cell numbers sharply decrease when meiosis is initiated. There is solid evidence (DNA ladders, in situdetection) that this loss is through apoptosis. Oocyte apoptosis appears to hit mitotic primordial germ cells (PGC), pachyteneoocytes and early primordial follicles. The control of oocyte apoptosis is not fully understood, although survival factors (LIF, kitligand and FGF), as well as death inducing factors (fas ligand, TGF!), have been identified. Fas ligand binding on oocytic fasmay result in caspase 8 activation. Two pathways inducing oocyte apoptosis may then be operating. In the first one, activatedcaspase 8 will induce activation of executioner caspases. In the second one, activated caspase 8 will trigger the cleavage of the bcl2family member Bid, which will act on mitochondria, resulting in cytochrome c release, caspase 9 activation and finally, activationof all executioner caspases. As a consequence of caspase activation, alterations in the cell nucleus (DNAse activation, PARPfragmentation), in the cell cytoskeleton (lamin) and cell metabolism will occur, producing cell death. During folliculogenesis, germcell loss, owing to oocyte apoptosis, has been postulated within primordial and preantral follicles. Its regulatory mechanisms maybe even more complex than those operating in foetal oocytes since additional control factors include EGF/TGF" and bcl2(survival) and activin (death inducer). In contrast, oocytes from antral follicles appear to be very unsensitive to death inducingstimuli. © 2000 Elsevier Science Ireland Ltd. All rights reserved.
Keywords: Oocyte; Apoptosis; Follicle
www.elsevier.com/locate/mce
1. Introduction
Females of all domestic species and primates have afinite store of germ cells, which is established duringoogenesis and used later for folliculogenesis. Formationof this store (i.e. the pool of primordial follicles) gener-ally occurs during foetal life as a result of a number ofoverlapping events, migration of the primordial germcells (PGC) to the gonadal ridge, proliferation of thesePGC, initiation of meiotic prophase followed by itsblock to the diplotene stage, and finally, formation ofthe primordial follicles, when cells originating from therete ovarii surround the oocytes. Germ cell loss appearshowever to occur at each of these steps. The first
section of this review will attempt to clarify how andwhy germ cell loss occurs throughout oogenesis.
Ewes and women ovulate every 17 and 28 days,respectively. This infrequent occurrence of ovulationcontrasts with the continuous exit from the reserve ofprimordial follicles (2 and 10–30 follicles initiategrowth) every day in sheep and in young women,respectively, according to Driancourt et al. (1985),Faddy and Gosden (1995). This suggests that a verystrong tuning of the number of growing follicles occursthroughout folliculogenesis. The respective contributionof oocyte apoptosis versus apoptosis of somatic cells togenerate a single ovulatory follicle from a continuousflux of growing follicles will be detailed in the secondsection of this review. For clarity, only three specieswhich differ in the timings of their phases of oogenesis(mouse versus sheep and humans) or in their strategiesto use their pool of primordial follicles for folliculogen-esis (mouse and sheep versus humans who displaymenopause) will be considered.
* Corresponding author. Tel.: +33-2-41228265; fax: +33-2-41228251.
E-mail address: [email protected](M.A. Driancourt).
0303-7207/00/$ - see front matter © 2000 Elsevier Science Ireland Ltd. All rights reserved.PII: S0303 -7207 (99 )00246 -4

Barres and Raff
Axonal Control of Oligodendrocyte Development
1125
as oligodendrocytes based on expression of characteristicantigenic markers. P8 optic nerves have about 35,000 oli-godendrocytes per nerve; when examined 10 d after a P8transection on P18, the nerves contain only about 3,000oligodendrocytes, compared with the 125,000 found incontrol P18 nerves (Barres et al., 1993b). Thus not only arefew new oligodendrocytes generated after a P8 transec-tion, but
.
90% of the oligodendrocytes present at P8 die.The death of oligodendrocytes after P8 transection is pre-vented if the levels of IGF-1, CNTF (Barres et al., 1993b),or neuregulins (NRG; P.-A. Fernandez, D. Tang, L.Cheng, A . Mudge, and M. Raff, manuscript submitted forpublication) are experimentally elevated.
How do axons regulate oligodendrocyte survival? Oli-godendrocytes do not die if the optic nerve is transected inWLD mutant mice in which the axons do not degenerate(Brown et al., 1991) and the ability of axons to promoteoligodendrocyte survival does not depend on electrical ac-tivity in the axons (Barres et al., 1993b). Purified neurons,but not neuron-conditioned culture medium, promote thesurvival of purified oligodendrocytes in vitro (Barres et al.,1993b). These findings suggest that the axon-derived sig-nal is contact-mediated and not dependent on electricalactivity. Neuregulins have recently been proposed to besuch a signal (see below).
How does one reconcile the findings that both astrocyte-derived and axon-derived signals seem to promote oligo-dendrocyte survival? It is possible that signals from bothsources collaborate to promote the survival of oligoden-drocytes; alternatively, newly formed oligodendrocytesmay depend on the astrocyte-derived signals while moremature oligodendrocytes may lose their dependence onastrocytes and come to depend solely on axons for theirsurvival.
Immature Schwann cells are also strongly dependent onaxons for a survival signal (Ciutat et al., 1996; Grinspanet al., 1996; Syroid et al., 1996; Trachtenberg and Thomp-son, 1996; Carroll et al., 1997; Nakao et al., 1997). Interest-ingly, one week after PNS myelination, Schwann cells nolonger depend on axons for their survival (Grinspan etal., 1996) and may instead depend on autocrine signals(Cheng et al., 1998). The same is true for oligodendro-cytes, many but not all of which survive transection inadult optic nerves (Vaughn and Pease, 1970; Fulcrand andPrivat, 1977; McPhilemy et al., 1990; Ludwin, 1990). Thesechanges in survival requirements are reminiscent of thechanges that occur in a number of types of neurons, in-cluding sensory DRG neurons and sympathetic neurons,that initially depend on target-derived signals for survivalbut lose this dependence in the adult (Acheson et al.,1995).
A Model for How Axons ControlOligodendrocyte Number
A tentative model for how a competition for axon-depen-dent survival signals may help to match oligodendrocyteand axon numbers during development has been proposed(Barres et al., 1993b; Barres and Raff, 1994). Once an oli-godendrocyte precursor cell stops dividing and begins todifferentiate into an oligodendrocyte, its specific require-ments for survival signals change: it rapidly loses its PDGF
receptors, for example, so that PDGF can no longer pro-mote its survival (Hart et al., 1989; McKinnon et al., 1990).It now has only 2–3 d to contact a nonmyelinated region ofaxon that provides new signals that are required for itscontinued survival. A cell that fails to find an axon will killitself (Fig. 1). Forcing oligodendrocytes to compete foraxon-dependent survival signals that are limited in amountor availability would help to ensure that the final numberof oligodendrocytes is precisely matched to the number(and length) of axons requiring myelination. Importantly,according to this model, newly formed premyelinating oli-godendrocytes depend on astrocyte-derived signals fortheir survival for about the first 2 d, whereas after 3 d theoligodendrocytes are more mature and depend primarilyupon an axon-derived signal.
Much evidence supports such a model. The model ex-plains why most developing oligodendrocytes that die doso 2 to 3 d after their generation and why most developingoligodendrocytes die after axotomy. It also explains whythere appears to be a perfect matching between oligoden-drocytes in the optic nerve and the number and lengths ofaxons (see Barres and Raff, 1994); in normal CNS whitematter, all mature oligodendrocytes that survive seem tomyelinate axons. Since the proposal of this model, severalof its predictions have been tested. One prediction is thatif the number of axons is experimentally increased, thenthe number of oligodendrocytes that survive will increaseproportionally. This has been found to be the case (Burneet al., 1996). Another prediction is that oligodendrocytesthat succeed in contacting axons will preferentially sur-vive, while those that don’t will die. This has been testedand found to be true (Trapp et al., 1997). A last predictionis that if the number of oligodendrocytes generated is ex-
Figure 1. A model for how oligodendrocyte number is matchedto axonal surface area. Once an OPC stops dividing and differen-tiates into an oligodendrocyte (left side of figure), it has 2–3 d tocontact an unmyelinated region of axon, which provides a newsignal that the cell requires for continued survival. A strocyte-derived signals, such as PDGF, can promote the survival of newlyformed oligodendrocytes for at least 2 d (middle of figure). Butas the newly formed oligodendrocytes undergo further matura-tion (right side of figure), they lose responsiveness to these astro-cyte-derived signals and require an axonal signal to survive.Those that fail to contact an axon by 3 d after generation undergoapoptosis.
on Decem
ber 21, 2013jcb.rupress.org
Dow
nloaded from
Published December 13, 1999
3. regulating cell number: apoptosis in neural cells
h]p://3.bp.blogspot.com/_kaQ5P19FVgk/SfYRJU7lyPI/AAAAAAAAC2g/X1Hdc8y4Wo4/s400/Oligodendrocyte1.JPG

Cell114
a consequence of the sensing of competitive stress. Anintriguing question that remains is how cells are able tosense competition. One possibility is that cells competefor sufficient levels of a survival factor that normallyblocks hid expression. Dpp signaling promotes cell sur-vival in the wing disc (Moreno et al., 2002a) but appearsto be unaffected in discs expressing dMyc. Alternatively,some cells in competition may be deprived of adequatenutrients, although in our experiments, cells at a growthdisadvantage retain a normal nucleolar size, arguing thattheir biosynthetic rates are not abnormally low. However,our results suggest that dMyc provokes competition andhid expression via a short-range signal, since close prox-imity is required for the perception of competitive effects.Perhaps the most intriguing feature of this signal is thatit is not perceived by nearby cells across a compartmentboundary, although dMyc induces competition betweencells within the posterior compartment as well as withinthe anterior (data not shown). One possibility is thatcells expressing dMyc acquire adhesive properties that
Figure 5. Cell Competition and Apoptosis as Components of Organtransmit a competitive signal to neighboring cells, whichSize Controlis not compatible with the adhesive barrier that main-(A) During cell competition, cells with a growth advantage (green)tains the compartment boundary.signal to wild-type cells (gray), inducing the expression of the pro-apoptotic gene hid and eventually their death. During subsequent
Growth Requirements for Cell Competition growth, cell competition continues until the fast-growing cells haveOur studies reveal that cell competition is not invariably populated most of the organ while eliminating most of the wild-type
cells. Due to elimination of cells through apoptosis, normal andinduced whenever rapidly growing cells populate re-uniform organ size is reached at the end of the development.gions of a developing organ. Both the PI3K Dp110 and(B) When apoptosis is blocked by genetic elimination of hid functioncyclin D/Cdk4 potently promote growth when overex-or ubiquitous expression of P35, wild-type (gray) cells populatepressed, yet they do not induce competition in any ofportions of the organ, but a wide variation of final organ sizes is
our assays. These observations also demonstrate that observed. Thus, elimination of apoptosis prevents uniformity of or-balanced growth—growth that simultaneously drives gan size.cell division and cellular growth—is not required to in-duce cell competition. dMyc expression increasesclonal mass solely by increasing cell size (Johnston et petition and leads to overgrowth of the compartment inal., 1999). Thus, this trait of cell competition may be which the dMyc-expressing cells reside.related to a size-measuring mechanism that recognizes An important conclusion of this work is that apoptosistotal mass rather than cell number (Neufeld et al., 1998). is critical for appropriate wing development. Our experi-However, Dp110 also promotes growth primarily by in- ments demonstrate that apoptosis has two roles in regu-creasing cell size, indicating that qualitative differences lating wing size. One role is uncovered when the discexist in the cellular response to expression of dMyc is challenged by local changes in dMyc levels, condi-and Dp110. Although both growth regulators increase tions in which cells are exceptionally sensitive to hidprotein synthesis, dMyc probably does so by increasing gene dosage: the full hid complement is necessary forcomponents of the protein synthetic machinery (initia- the disc to respond to competition properly and elimi-tion factors and ribosomal proteins, etc.) (Orian et al., nate cells. However, a second role of apoptosis is re-2003), whereas PI3K signaling is thought to function by vealed when it is abolished: this role regulates uniformityincreasing the utilization of existing machinery (Stocker of disc size, and its loss is manifested as a widening ofand Hafen, 2000; Thomas, 2000). Regardless of the the range of disc sizes within a given population. Thismechanism, our experiments argue against the notion second role of apoptosis indicates that organ over-that apposed populations of fast- and slow-growing growth is distinct from loss of organ size control. Wingcells always result in cell competition. overgrowth—observed when cell competition is not ex-
ecuted during local growth perturbations—occurs suchthat, although larger than normal, wing size still fallsAppropriate Size Control Requires Apoptosis
We have provided three lines of evidence that indicate within a uniform range. In contrast, loss of size controlis the absence of a discrete and reproducible size popu-that cell competition leading to cell death is required
for control of wing size. First, growth induced by local lation and results from a failure to induce apoptosisduring the process of growth (Figure 5). Based on ourexpression of either Dp110 or cyclin D ! Cdk4 does
not induce competition and causes wing overgrowth. observations, we propose that hid-regulated apoptosiscontributes to a disc-intrinsic mechanism that limitsSecond, when dMyc is expressed in all cells of the wing
disc, the wing overgrows, whereas the introduction of variation in size by allowing elimination of cells. Thismechanism may serve as negative feedback to the posi-clones lacking dMyc leads to cell competition and to
wings approaching normal size. Finally, genetic reduc- tive aspects of growth during development. Loss offeedback control could allow stochastic variation in size,tion of hid prevents the cell death associated with com-
3. regulating cell number: organ size & shape control

h]p://www.pilonidal.org/_assets/contentImages/woondHealing_phases_of_cutaneous.gif
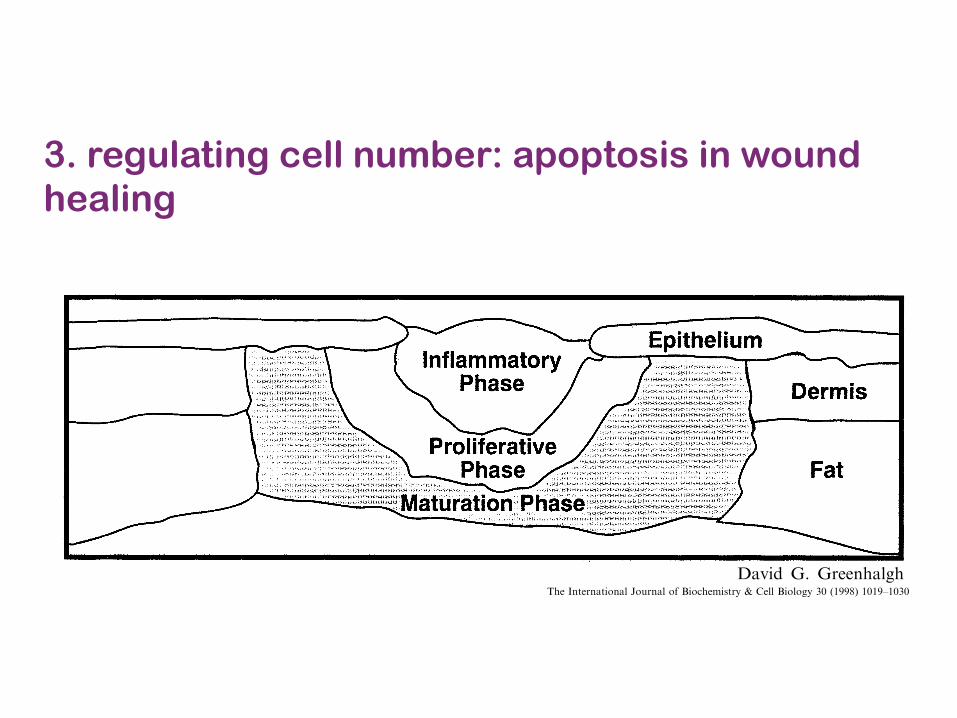
3. regulating cell number: apoptosis in wound healing
!"#$ %"&'() *#+(&", #-. +/(&01-!,2 3)!/ #$,&)#4! # "&$! 1- ()! 5)#%&+/(&,1, &* -&-41#6$! 7#8(!"1#$,2 9&&- #*(!" ()! #""14#$ &* 7#+"&5)#%!,()!"! 1, #- 1-:;< &* $/75)&+/(!,2 3)! "&$! &*$/75)&+/(!, 1- (1,,;! "!5#1" )#, #$,& 6!!- 1-4!,(18%#(!. 6/ !$171-#(1-% ()! +!$$, =>? #-. 6/ 1-4!,(18%#(1-% )!#$1-% 1- 71+! $#+01-% 5&5;$#(1&-, &*$/75)&+/(!, @,;+) #, A;.! 71+!B =C?2 3)! #+(;#$"&$! &* $/75)&+/(!, 1, -&( (&(#$$/ +$!#"D 6;( $/785)&+/(!, "!$!#,! 7#-/ +/(&01-!, ()#( 1-:;!-+!*;-+(1&- &* 7#+"&5)#%!, 1- #..1(1&- (& 7#-/&()!" +!$$,2 3)! 1-:#77#(&"/ "!#+(1&- 1-.;+!.6/ $!;0&+/(!, 1, !,,!-(1#$ *&" 5"!5#"1-% ()!'&;-. *&" ()! 5"&.;+(1&- &* # -!' !<("#+!$$;$#"7#("1< ()#( 7#1-$/ +&-,1,(, &* +&$$#%!-, #-. %$/8+&,#71-&%$/+#-,2 E- #- 1-+1,1&-D ()1, F!"#$%%$&'()*G () F+$,G -.$/0 $#,(, *&" *&;" (& H4! .#/,2 3)!(!"7 F$#%G 5)#,! 1, .!"14!. *"&7 ()! *#+( ()#(()!"! 1, 4!"/ $1(($! (!-,1$! ,("!-%() ,1-+! 1-1(1#$$/&-$/ H6"1- #-. H6"&-!+(1- #"! )&$.1-% ()! '&;-.(&%!()!"2
I/(&01-!, #-. %"&'() *#+(&", "!$!#,!. 6/ ()!1-:#77#(&"/ +!$$, #(("#+( H6"&6$#,(, #-. 7/&H86"&6$#,(, 1-(& ()! '&;-. (& 1-1(1#(! ()! F-)(+!10)$&'!20 () 3(++$,0" -.$/0G &* (1,,;! "!5#1"2 3)!"! 1, #"#51. 1-:;< &* H6"&6$#,(, #( JK> .#/, *&$$&'!.6/ ()! H",( .!5&,1(1&- &* +&$$#%!- #-. %$/+&,#718-&%$/+#-,2 L;"1-% ()1, 5)#,! ()!"! 1, # "#51.1-+"!#,! 1- ()! #7&;-(, &* +&$$#%!- .!(!+(!. 1-()! '&;-. ()#( 1, #,,&+1#(!. '1() # "#51.1-+"!#,! 1- 1-+1,1&-#$ (!-,1$! ,("!-%()2 M16"&6$#,(,"!N;1"! &</%!- #-. -;("1!-(, (& 7#-;*#+(;"! ()!!<("#+!$$;$#" 7#("1< ,& ()!"! 1, # "!N;1"!7!-( *&"
#-%1&%!-!,1,2 O#+"&5)#%!, 1- ()! )/5&<1+ !-841"&-7!-( "!$!#,! #-%1&%!-1+ *#+(&", ()#( ,(17;8$#(! 5&,(84!-;$! !-.&()!$1#$ +!$$, (& 6"!#0 *"!! &*()!1" 6#,!7!-( 7!76"#-! #-. 71%"#(! 1-(& ()!'&;-. (& +"!#(! # )1%)$/ 4#,+;$#" F%"#-;$#(1&-(1,,;!G =P?2 3)1, !-41"&-7!-( &* H6"&6$#,(,D -!'+#51$$#"1!, #-. !<("#+!$$;$#" 7#("1< +"!#(! #- #.!8N;#(! 6!. *&" ()! .1,";5(!. !51()!$1;7 (& 71%"#(!#+"&,, ()! '&;-.2 3)! +&$$#%!- 5"&.;+!. 1,!,,!-(1#$ *&" ()! '&;-. (& "!%#1- 7;+) &* ()!,("!-%() $&,( #*(!" 1-Q;"/2
R4!-(;#$$/D @;,;#$$/ SKJ '!!0, 1- #- 1-+1,1&-B+&$$#%!- +&-(!-( 1- ()! '&;-. $!4!$, &TD 7#"01-%()! ,(#"( &* ()! H-#$ 5)#,! &* (1,,;! "!5#1"U ()!F%$'4)$'!(" -.$/0G2 I&$$#%!- 1, ,(1$$ ,/-()!,1V!.6;( +&$$#%!-#,! 5"&.;+(1&- #$,& 1-+"!#,!, (& +"!8#(! # 6#$#-+! 6!('!!- +&$$#%!- 5"&.;+(1&- #-..!%"#.#(1&-2 L;"1-% ()1, 5)#,!D +&$$#%!- 1, "!7&8.!$!. #-. "!8#$1%-!. #$&-% 5$#-!, &* ,("!,, (&1-+"!#,! ()! ,("!-%() &* ()! 1-+1,1&-2 W&;-.,)#4! 5!",1,(!-( 1-+"!#,!, 1- 4#,+;$#"1(/ #-. +!$$;8$#"1(/ ()#( 5!",1,(, *&" 7&-(), #, 1-.1+#(!. 6/ ()!+&-(1-;!. 5"!,!-+! &* # "!. #-. ()1+0!-!.'&;-.2 R4!-(;#$$/D ()! '&;-. 6!+&7!, $!,, +!$8$;$#" #-. 4#,+;$#" (& !-. ;5 '1() # '&;-. ()#(1, :#( #-. ')1(!2 X,;#$$/ ()! 7#(;"#(1&- 5)#,!$#,(, #55"&<17#(!$/ &-! /!#" 6!*&"! ()! '&;-..!4!$&5, 1(, H-#$ ,(#(! &* # 7#(;"! #4#,+;$#" #-.#+!$$;$#" ,+#"2
!" #$%&$'&()* &) '+, -,../.$% 0$'',%) $1',% &)2/%3
O&,( &* ()! 1-*&"7#(1&- "!%#".1-% ()! 5)#,!,&* (1,,;! "!5#1" '!"! .!(!"71-!. *&" 1-+1,1&-#$'&;-.,2 E- &()!" (/5!, &* '&;-.,D !,5!+1#$$/&5!- '&;-.,D ()! 5)#,!, &* (1,,;! "!5#1" (!-. (&6$!-. (&%!()!" #-. +#- +&8!<1,( 1- .1T!"!-( $&8+#(1&-, .!5!-.!-( &- ()! .!%"!! &* (1,,;! "!5#1"2Y ,5!+(";7 &* ()! 4#"1&;, 5)#,!, !<1,(, '1() ()!1-:#77#(&"/ 5)#,! 6!1-% *&;-. 1- ()! +!-("#$&5!- #"!#, ')1$! 7&"! 7#(;"! 5)#,!, #"! *&;-.#, &-! ("#4!$, (&'#"., ()! &"1%1-#$ '&;-. !.%!@M1%2 SB2 3)!"! 1, 5!",1,(!-( 1-:#77#(1&- 1- ()!!<5&,!. +!-(!" &* ()! '&;-. 6!+#;,! ()!"! 1, #+&-(1-;#$ 1-4#,1&- &* 71+"&8&"%#-1,7, #-.*&"!1%- 7#(!"1#$ ()#( #+( #, 1-:#77#(&"/ ,(17;$12E-:#77#(1&- '1$$ 5!",1,( #, $&-% #, ()! '&;-.
M1%2 S2 E- #- &5!- '&;-.D #$$ ()"!! 5)#,!, +&8!<1,( (&%!()!"2E- ()! +!-(!"D ()! !<5&,!. '&;-. 1, +&-(1-;#$$/ ,(17;$#(!. (&5"&.;+! 1-:#77#(&"/ 7!.1#(&", #-. )#, # 5"&$&-%!. 1-:#787#(&"/ 5)#,! &* )!#$1-% ;-(1$ ()! '&;-. 1, +&4!"!. 6/ #- !518()!$1;72 O&"! #.4#-+!. 5)#,!, 5"&%"!,, 6!-!#() ()!!51()!$1;7 (&'#". ()! &"1%1-#$ '&;-. !.%!2
5676 7)00".$+,. 8 9.0 :"'0)"$'!("$+ ;(4)"$+ (1 <!(3.0%!/')* = >0++ <!(+(,* ?@ ABCCDE B@BCFB@?@ Z[SZ
!"# $%&# %' ()%)*%+,+ ,- .%/-0 "#(&,-1
2(3,0 45 4$##-"(&1" 6
!"#$%&'"(& )* +,%-"%./ +0%1("%2 3)2#1&$42 *)% 50146%"(/ 7)%&0"%( 5$41*)%(1$ $(6 8(19"%21&. )* 5$41*)%(1$/ !$912/ :;:< +&)=>&)(?),4"9$%6/ +$=%$'"(&)/ 5@ A<BCD/ 8+@
7#8#,3#0 9: ;(-/($< :==>? (88#)*#0 @ A(< :==>
!"#$%&'$
B%/-0 "#(&,-1 ,-3%&3#+ ( +#$,#+ %' $(),0 ,-8$#(+#+ ,- +)#8,C8 8#&& )%)/&(*,%-+ *"(* )$#)($# *"# .%/-0 '%$ $#)(,$D0#)%+,* -#. E(*$,8#+ (-0 C-(&&<D E(*/$# *"# .%/-05 F)%- 8%E)&#*,-1 *"#,$ *(+G+D *"#+# +)#8,C8 8#&& *<)#+ E/+* H##&,E,-(*#0 '$%E *"# .%/-0 )$,%$ *% *"# )$%1$#++,%- *% *"# -#I* )"(+# %' "#(&,-15 !"# E%+* &%1,8(& E#*"%0 %'8#&&/&($ 0%.-J$#1/&(*,%- ,+ *"$%/1" ()%)*%+,+5 K)%)*%+,+ (&&%.+ '%$ *"# #&,E,-(*,%-+ %' #-*,$# )%)/&(*,%-+ .,*"%/**,++/# 0(E(1# %$ (- ,-L(EE(*%$< $#+)%-+#5 !",+ $#3,#. 0,+8/++#+ .",8" 8#&&+ 0%E,-(*# *"# 3($,%/+ )"(+#+ %' *,++/#$#)(,$ (-0 "%. *"# 8#&&/&($ )(**#$- E(< 3($< ('*#$ 0,M#$,-1 *<)#+ %' ,-N/$<5 !"# )%*#-*,(& E#8"(-,+E+ ,-3%&3#0 ,-*"# 0%.-J$#1/&(*,%- %' ,-L(EE(*,%- (-0 CH$%+,+ ($# (&+% 8%3#$#05 !"# +*/0,#+ *"(* +/))%$* *"# "<)%*"#+,+ *"(*()%)*%+,+ ,+ ,-3%&3#0 ,- *"# $#1/&(*,%- %' .%/-0 "#(&,-1 ($# 0,+8/++#05 !"# #3,0#-8# +/))%$*,-1 )%*#-*,(& 8#&& +,1-(&+,-3%&3#0 ,- *"# ,-0/8*,%- %' ()%)*%+,+ ,- *,++/# $#)(,$ ($# #I(E,-#05 O,-(&&<D *"# $#3,#. #-0+ .,*" ( )$#+#-*(*,%- %'"%. 0<+$#1/&(*,%- %' ()%)*%+,+ 8(- &#(0 *% )(*"%&%1,8 '%$E+ %' "#(&,-1 +/8" (+ #I8#++,3# +8($$,-1 (-0 CH$%+,+5 P</-0#$+*(-0,-1 *"# E#8"(-,+E+ 8%-*$%&&,-1 ()%)*%+,+ (-0 *,++/# $#)(,$D %-# E(< #3#-*/(&&< 0#3#&%) *"#$()#/*,8E%0(&,*,#+ *% E,-,E,Q# +8($$,-1D ( C-(& )(*".(< '%$ E(-< 0,+#(+# )$%8#++#+5 ! :==> R&+#3,#$ S8,#-8# T*05 K&& $,1"*+$#+#$3#05
E".F)%62G K)%)*%+,+? B%/-0 "#(&,-1? 4$%.*" '(8*%$+? S,1-(& *$(-+0/8*,%-
U' *"#$# ,+ 0(E(1# *% %-#V+ "%/+#D *"# %.-#$W/,8G&< 8(&&+ ( +#$,#+ %' 0,M#$#-* $#)(,$ )#%)($)#-*#$+D #*$,8,(-+D )&/EH#$+D )(,-*#$+Y ."%#(8" "(3# *% *(G# */$-+ ,- 0%,-1 *"#,$ N%H+ *%$#)(,$ *"# 0(E(1#5 !"# %.-#$ .(-*+ *"# .%$G#$+*% ($$,3# W/,8G&<D 0% *"#,$ N%H+ (-0 *"#- &#(3# +%*"(* *"# -#I* )"(+# %' $#8%-+*$/8*,%- 8(- %88/$5K-< 1$%/) *"(* +*(<+ *%% &%-1 #-0+ /) H#,-1 (-,$$,*(*,%- (-0 0#&(<+ *"# /&*,E(*# %/*8%E#5 U- (+,E,&($ .(<D *,++/# $#)(,$ ,-3%&3#+ ( +#$,#+ %'
#3#-*+ *"(* ,-3%&3# $(),0 ,-8$#(+#+ ,- +)#8,C8 8#&&)%)/&(*,%-+ *% )#$'%$E ( +)#8,C8 *(+G +/8" (+ ,-JL(EE(*,%- %$ 8%&&(1#- 0#)%+,*,%-5 B"#- #(8"#3#-* ,+ 8%E)&#*#0 *"# #M#8*%$ 8#&&+ E/+* &#(3#*"# .%/-0 *% (&&%. '%$ *"# +*($* %' *"# -#I*#3#-* ,- .%/-0 "#(&,-15 R(8" +/H+#W/#-* )"(+#,-3%&3#+ ,-8$#(+#+ ,- %*"#$ 8#&& )%)/&(*,%-+ *"(*H< -#8#++,*< "(3# *% 1$(0/(&&< 0#8$#(+# ,- -/EJH#$5 R3#-*/(&&<D ( E(*/$# .%/-0 #-0+ /) W/,*#(8#&&/&($ (-0 (3(+8/&($5 !"# 8#&&+ *"(* 0,+())#($'$%E ( .%/-0 8(- &#(3# H< %-# %' *"$## E#8"(-J,+E+Z -#8$%+,+D #E,1$(*,%- %$ ()%)*%+,+5 7#8#-*#3,0#-8# +/11#+*+ *"(* ()%)*%+,+ ,+ *"# E(,-8(/+# %' 0#8$#(+,-1 8#&&/&($,*< 0/$,-1 *"# 3($,%/+
!"# U-*#$-(*,%-(& ;%/$-(& %' P,%8"#E,+*$< [ \#&& P,%&%1< ]^ X:==>Y :^:=_:^]^
:]@`J9`9@a=>ab:=5^^ ! :==> R&+#3,#$ S8,#-8# T*05 K&& $,1"*+ $#+#$3#05cUUZ S:]@`J9`9@X=> Y^^^@>J9
!"#$%&#'%(&)*%(+,*-'%(+ *.
/)*0"#1)2&'34 5#++ /)*+*63
()*+!,-.
6 \%$$#+)%-0,-1 (/*"%$5 !#&5Z d:J=:eJf@]9^@^? O(IZ d:J=:eJf@]9]`]? RJE(,&Z 01"(&1"gW/,G-#*58%E
!"# $%&# %' ()%)*%+,+ ,- .%/-0 "#(&,-1
2(3,0 45 4$##-"(&1" 6
!"#$%&'"(& )* +,%-"%./ +0%1("%2 3)2#1&$42 *)% 50146%"(/ 7)%&0"%( 5$41*)%(1$ $(6 8(19"%21&. )* 5$41*)%(1$/ !$912/ :;:< +&)=>&)(?),4"9$%6/ +$=%$'"(&)/ 5@ A<BCD/ 8+@
7#8#,3#0 9: ;(-/($< :==>? (88#)*#0 @ A(< :==>
!"#$%&'$
B%/-0 "#(&,-1 ,-3%&3#+ ( +#$,#+ %' $(),0 ,-8$#(+#+ ,- +)#8,C8 8#&& )%)/&(*,%-+ *"(* )$#)($# *"# .%/-0 '%$ $#)(,$D0#)%+,* -#. E(*$,8#+ (-0 C-(&&<D E(*/$# *"# .%/-05 F)%- 8%E)&#*,-1 *"#,$ *(+G+D *"#+# +)#8,C8 8#&& *<)#+ E/+* H##&,E,-(*#0 '$%E *"# .%/-0 )$,%$ *% *"# )$%1$#++,%- *% *"# -#I* )"(+# %' "#(&,-15 !"# E%+* &%1,8(& E#*"%0 %'8#&&/&($ 0%.-J$#1/&(*,%- ,+ *"$%/1" ()%)*%+,+5 K)%)*%+,+ (&&%.+ '%$ *"# #&,E,-(*,%-+ %' #-*,$# )%)/&(*,%-+ .,*"%/**,++/# 0(E(1# %$ (- ,-L(EE(*%$< $#+)%-+#5 !",+ $#3,#. 0,+8/++#+ .",8" 8#&&+ 0%E,-(*# *"# 3($,%/+ )"(+#+ %' *,++/#$#)(,$ (-0 "%. *"# 8#&&/&($ )(**#$- E(< 3($< ('*#$ 0,M#$,-1 *<)#+ %' ,-N/$<5 !"# )%*#-*,(& E#8"(-,+E+ ,-3%&3#0 ,-*"# 0%.-J$#1/&(*,%- %' ,-L(EE(*,%- (-0 CH$%+,+ ($# (&+% 8%3#$#05 !"# +*/0,#+ *"(* +/))%$* *"# "<)%*"#+,+ *"(*()%)*%+,+ ,+ ,-3%&3#0 ,- *"# $#1/&(*,%- %' .%/-0 "#(&,-1 ($# 0,+8/++#05 !"# #3,0#-8# +/))%$*,-1 )%*#-*,(& 8#&& +,1-(&+,-3%&3#0 ,- *"# ,-0/8*,%- %' ()%)*%+,+ ,- *,++/# $#)(,$ ($# #I(E,-#05 O,-(&&<D *"# $#3,#. #-0+ .,*" ( )$#+#-*(*,%- %'"%. 0<+$#1/&(*,%- %' ()%)*%+,+ 8(- &#(0 *% )(*"%&%1,8 '%$E+ %' "#(&,-1 +/8" (+ #I8#++,3# +8($$,-1 (-0 CH$%+,+5 P</-0#$+*(-0,-1 *"# E#8"(-,+E+ 8%-*$%&&,-1 ()%)*%+,+ (-0 *,++/# $#)(,$D %-# E(< #3#-*/(&&< 0#3#&%) *"#$()#/*,8E%0(&,*,#+ *% E,-,E,Q# +8($$,-1D ( C-(& )(*".(< '%$ E(-< 0,+#(+# )$%8#++#+5 ! :==> R&+#3,#$ S8,#-8# T*05 K&& $,1"*+$#+#$3#05
E".F)%62G K)%)*%+,+? B%/-0 "#(&,-1? 4$%.*" '(8*%$+? S,1-(& *$(-+0/8*,%-
U' *"#$# ,+ 0(E(1# *% %-#V+ "%/+#D *"# %.-#$W/,8G&< 8(&&+ ( +#$,#+ %' 0,M#$#-* $#)(,$ )#%)($)#-*#$+D #*$,8,(-+D )&/EH#$+D )(,-*#$+Y ."%#(8" "(3# *% *(G# */$-+ ,- 0%,-1 *"#,$ N%H+ *%$#)(,$ *"# 0(E(1#5 !"# %.-#$ .(-*+ *"# .%$G#$+*% ($$,3# W/,8G&<D 0% *"#,$ N%H+ (-0 *"#- &#(3# +%*"(* *"# -#I* )"(+# %' $#8%-+*$/8*,%- 8(- %88/$5K-< 1$%/) *"(* +*(<+ *%% &%-1 #-0+ /) H#,-1 (-,$$,*(*,%- (-0 0#&(<+ *"# /&*,E(*# %/*8%E#5 U- (+,E,&($ .(<D *,++/# $#)(,$ ,-3%&3#+ ( +#$,#+ %'
#3#-*+ *"(* ,-3%&3# $(),0 ,-8$#(+#+ ,- +)#8,C8 8#&&)%)/&(*,%-+ *% )#$'%$E ( +)#8,C8 *(+G +/8" (+ ,-JL(EE(*,%- %$ 8%&&(1#- 0#)%+,*,%-5 B"#- #(8"#3#-* ,+ 8%E)&#*#0 *"# #M#8*%$ 8#&&+ E/+* &#(3#*"# .%/-0 *% (&&%. '%$ *"# +*($* %' *"# -#I*#3#-* ,- .%/-0 "#(&,-15 R(8" +/H+#W/#-* )"(+#,-3%&3#+ ,-8$#(+#+ ,- %*"#$ 8#&& )%)/&(*,%-+ *"(*H< -#8#++,*< "(3# *% 1$(0/(&&< 0#8$#(+# ,- -/EJH#$5 R3#-*/(&&<D ( E(*/$# .%/-0 #-0+ /) W/,*#(8#&&/&($ (-0 (3(+8/&($5 !"# 8#&&+ *"(* 0,+())#($'$%E ( .%/-0 8(- &#(3# H< %-# %' *"$## E#8"(-J,+E+Z -#8$%+,+D #E,1$(*,%- %$ ()%)*%+,+5 7#8#-*#3,0#-8# +/11#+*+ *"(* ()%)*%+,+ ,+ *"# E(,-8(/+# %' 0#8$#(+,-1 8#&&/&($,*< 0/$,-1 *"# 3($,%/+
!"# U-*#$-(*,%-(& ;%/$-(& %' P,%8"#E,+*$< [ \#&& P,%&%1< ]^ X:==>Y :^:=_:^]^
:]@`J9`9@a=>ab:=5^^ ! :==> R&+#3,#$ S8,#-8# T*05 K&& $,1"*+ $#+#$3#05cUUZ S:]@`J9`9@X=> Y^^^@>J9
!"#$%&#'%(&)*%(+,*-'%(+ *.
/)*0"#1)2&'34 5#++ /)*+*63
()*+!,-.
6 \%$$#+)%-0,-1 (/*"%$5 !#&5Z d:J=:eJf@]9^@^? O(IZ d:J=:eJf@]9]`]? RJE(,&Z 01"(&1"gW/,G-#*58%E
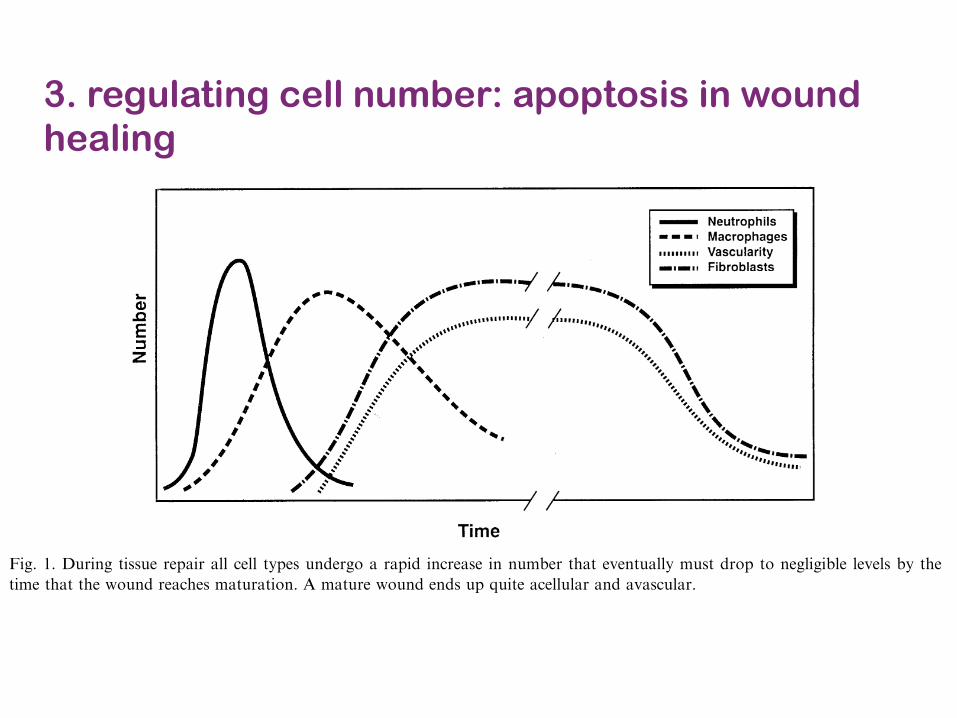
3. regulating cell number: apoptosis in wound healing
!"#$%! &' (%#)*+$, -.&."&!*! *! "(% /&!" )&$*0#)/%0(#+*!/ &' 0%)) %)*/*+#"*&+ !*+0% *" *! %+%1$2%30*%+" 40%))! 5& +&" (#6% "& /*$1#"%7 #+5 *" 5&%!+&" )%#5 "& *+8#//#"*&+, 9&"%+"*#) !&:10%! &'#.&."&!*! !*$+#)! (#6% #)!& ;%%+ *+6%!"*$#"%5 #+5(#6% ;%%+ '&:+5 "& &1*$*+#"% *+ "(% "*!!:% "(#"0&6%1! "(% &.%+ <&:+5= <(%"(%1 %.*"(%)*:/= !>*+$1#'" &1 8#., ?(%!% @+5*+$! @" <*"( "(% 0)*+*0#)%A.%1*%+0% "(#" !:$$%!"! "(#" <&:+5! "(#" 1%/#*+&.%+ (#6% # .%1!*!"%+" *+8#//#"&12 1%!.&+!%,B+0% "(% <&:+5 *! 0&6%1%5 <*"( %.*"(%)*:/ &1$1#'"= "(%+ "(% *+8#//#"&12 1%!.&+!% 1#.*5)21%!&)6%!, C*+#))2= "(% #0":#) !*$+#)! "(#" #1%*+6&)6%5 *+ "(% #.&."&!*! !*$+#)*+$ +%%5 "& ;%5%"%1/*+%5, D2 :+5%1!"#+5*+$ "(% 0%)) !*$+#)!"(#" #1% *+6&)6%5 *+ "(% 0&+"1&) &' 0%)) .&.:E)#"*&+! *+ (%#)*+$ <&:+5!= 0)*+*0*#+! 0&:)5 5%!*$+"1%#"/%+"! "(#" 0&:)5 (%). /*+*/*F% "(% 0#:!%! &'%A0%!!*6% *+8#//#"*&+ #+5 !0#11*+$,
!" #$%%&%'( )*'+,$- './$( 01&+2 *$'%3+,
-'"%1 *+G:12 # $1%#" 6#1*%"2 &' !*$+#)! #1%1%)%#!%5 "(#" +&" &+)2 *+*"*#"% ;:" 1%$:)#"% #))#!.%0"! &' "*!!:% 1%.#*1, C&1 5%0#5%!= (*!"&)&$*0#+#)2!%! 1%6%#)%5 "(#" "(%1% *! # 1%$:)#1 !%H:%+0%&' 5*I%1*+$ 0%)) .&.:)#"*&+! "(#" #..%#1%5 *+ #+*+0*!*&+#) <&:+5 4C*$, J7, ?(% 1&)% &' %#0( 5*I%1E
%+" 0%)) .&.:)#"*&+ (#! ;%%+ *+6%!"*$#"%5 #+5 0:1E1%+")2 # ':+0"*&+ '&1 /&!" "2.%! (#! ;%%+.1&.&!%5, ?& 5%"%1/*+% "(% ':+0"*&+ &' %#0(.&.:)#"*&+= !.%0*@0 0%)) "2.%! <%1% %)*/*+#"%5 "&5%"%1/*+% <(%"(%1 "*!!:% 1%.#*1 <#! #)"%1%5,9)#"%)%"! #11*6% #" "(% "*/% &' 6#!0:)#1 5*!1:."*&+#+5 (#6% ;%%+ !(&<+ "& 0&+"#*+ )#1$% #/&:+"!&' $1&<"( '#0"&1! *+ "(%*1 #).(# $1#+:)%!, K1&<"('#0"&1! 1%)%#!%5 ;2 .)#"%)%"! *+*"*#"% "(% "*!!:%1%.#*1 ;2 #""1#0"*+$ 0%))! *+"& "(% <&:+5,L)*/*+#"*&+ &' +%:"1&.(*)! 1%!:)"%5 *+ *+01%#!%5<&:+5 *+'%0"*&+ ;:" 5*5 +&" 1%!:)" *+ (%#)*+$#;+&1/#)*"*%! MJN, O:11%+")2= +%:"1&.(*)! #1% '%)""& ;% .1*/#1*)2 *+6&)6%5 *+ "(% @1!"E)*+% 5%'%+!%#$#*+!" *+6#5*+$ &1$#+*!/! #+5 "(% %)*/*+#"*&+&' '&1%*$+ /#"%1*#), ?(% &A2$%+ '1%%E1#5*0#)! #+5.1&"%#!%! 1%)%#!%5 ;2 "(% +%:"1&.(*)! #1% %I%0"*6%'&1 .%1'&1/*+$ "(% 5%'%+!*6% "#!>! ;:" "(%2 #)!�" #! P'1*%+5)2 @1%Q "& 5%!"1&2 6*#;)% "*!!:%! *'+&" )*/*"%5,
?(% +%A" 0%)) "(#" #..%#1! *+ # <&:+5 *! "(%/#01&.(#$%, ?(% 1&)% &' "(% /#01&.(#$% *+<&:+5 (%#)*+$ <#! *+6%!"*$#"%5 *+ JRST ;2U%*;&6*0( #+5 V&!! <(& %)*/*+#"%5 /#01&.(#$%!'1&/ "(% <&:+5 #+5 5*!0&6%1%5 "(#" <&:+5!(%#)%5 .&&1)2 MWN, X*+0% "(%+= !%6%1#) *+6%!"*$#"&1!(#6% 0&+0):5%5 "(#" "(% /#01&.(#$% *! # >%2&10(%!"1#"&1 &' "*!!:% 1%.#*1 MY= ZN, [#01&.(#$%!1%$:)#"% "*!!:% 1%.#*1 "(1&:$( "(% 1%)%#!% &' !%6E
C*$, J, \:1*+$ "*!!:% 1%.#*1 #)) 0%)) "2.%! :+5%1$& # 1#.*5 *+01%#!% *+ +:/;%1 "(#" %6%+":#))2 /:!" 51&. "& +%$)*$*;)% )%6%)! ;2 "(%"*/% "(#" "(% <&:+5 1%#0(%! /#":1#"*&+, - /#":1% <&:+5 %+5! :. H:*"% #0%)):)#1 #+5 #6#!0:)#1,
!"#" #$%%&'()*' + ,'% -&.%$&(./0&() 102$&() 03 4/05'%6/7.$8 9 :%)) 4/0)0*8 ;< =>??@A ><>?B><;<J]W]
3. regulating cell number: apoptosis in wound healing

Nature Reviews | Cancer
Suboptimal but viable cell in a homotypic environment +
Suboptimal cell Survival
a
Suboptimal cell eliminated bycell competition+
Death
b
A
A WT
WT cell eliminated in a supercompetitor background+
Supercompetitor cells
c WT
Nature Reviews | Cancer
Winner cell Loser cell
High survival signallingGrowth and proliferation
Low survivalsignalling
JNK Hid
Apoptosis
DPPDPP DPP
DPP
DPP
?
more efficiently acquiring and/or internal-izing extracellular ligands8,10. Nevertheless, it seems that increased rates of endocytosis are crucial10. In its simplest form, the ligand-capture hypothesis assumes that the ligand is in limiting supply and that it represents a survival signal; from these two conjectures it follows that its partial withdrawal triggers apoptosis. This simplistic model resembles the trophic interactions previously described for neurons in peripheral tissues38.
However, some evidence suggests that the ligands that cells compete for are not in limit-ing supply. For example, cells near the source of DPP production experience surprisingly strong competition despite the high concen-tration of ligand available8. If the ligands for which cells compete are not limiting, another system would have to collaborate in order to discriminate different signalling levels. For example, a cell–cell communication system would have to ensure that cells can inform — and kill — each other based on the relative degree of ligand transduction. We do not know how DPP signalling levels are compared between neighbouring cells, but a relative reduction of DPP signalling can acti-vate the stress-inducible JNK pathway8,39,40 that ultimately triggers the execution of the apoptotic programme by activating the tran-scription of the pro-apoptotic gene Hid (also known as W)41,42. The requirement for JNK signalling for cell competition has been dis-puted9,43, however, and it has been suggested that only Hid, and not jnk, is required for the induction of apoptosis9. In addition, it has
been proposed that cells completely lacking the ability to transduce the DPP signal may be able to survive44,45, suggesting that other factors or nutrients — or even competition for attachment points at the extracellular matrix — might also be competed for and might contribute to the elimination of loser cells10,27,32. Alternatively, the juxtaposition of cells with high and low DPP signalling lev-els may be more important in the induction of apoptosis than absolute levels8,10,31,38–40,46. Although current research suggests that DPP is a vital factor for which cells struggle during competition (FIG. 3), further research is required to fully clarify the connections between DPP signalling and cell survival.
Cell engulfment. Winning cells appear to also have a final weapon against the losing cells. Previous work in Caenorhabditis elegans has shown that engulfment cooperates with apoptosis when the apoptotic signal is weak47,48. Cells homozygous for a hypomorphic allele of the caspase gene ced-3, which have weak-ened caspase activity, were engulfed and killed by surrounding cells, but survived if engulfment was prevented. Recent results in D. melanogaster indicate an even greater role of engulfment during cell competition11; engulfment must cooperate with a cell-death programme that is insufficient to remove loser cells by itself. The data suggest that both diminished survival signalling in the loser cells and activated engulfment in the win-ner cells occur and are necessary in parallel
to kill loser cells at the interface between winners and losers. Using mutations in several genes required for engulfment, it has been shown that cell competition cannot occur without engulfment and it has been proposed that winner cells require engulf-ment genes to “eat their way through mosaic tissues”, consuming out-competed cells along the way11. The genes examined in this context were draper (drpr, which encodes a transmembrane receptor that is related to the C. elegans engulfment receptor CED-1 (REFS 49,50) and which is required for phagocytosis by cultured D. melanogaster cells)51, WASp (which is an actin regulator also required for phagocytosis by cultured D. melanogaster cells52; its mammalian homologue is essential for phagocytosis by macrophages and neutrophils53,54) and the phosphatidylserine receptor (PSR, which has a more controversial role in engulf-ment55,56). All in all, the current model proposes that cell competition requires both survival signalling and engulfment: after the initial events trigger a reduction in survival signalling and a weak apoptotic signal in the loser cells, an active programme of engulfment is activated in the winner cells, crucially contributing to the killing of the out-competed cells11. If the survival signal-ling deficit of the loser cells is the initial event, then the specific signals that are sent by the loser cells to activate the engulfment machinery in the winner cells remain to be determined.
Figure 2 | A simple but stringent definition for cell competition-mediated apoptosis. a | A clone of cells with mutant genotype A is viable when situated among cells from the same genotype. b | However, cells with mutant genotype A are culled by apoptosis when situated among wild-type (WT) cells. During cell competition, WT cells are always the winners, and this could be a mechanism of maximizing tissue fitness and optimizing organ function by eliminating suboptimal cells. c | By contrast, in the case of super-competition, mutant cells are able to eliminate WT cells. Super-competition could contribute to cancer progression before morphological malformations are detectable.
Figure 3 | The ligand capture hypothesis. Cells compete for extracellular survival and growth factors such as decapentaplegic (DPP). As in a Darwinian competition, winner cells are able to obtain more survival factors. Loser cells, with low survival signalling as a consequence of the com-petition, activate the JUN N-terminal kinase (JNK) pathway and/or Hid transcription, which leads to apoptosis. Comparison of the DPP signalling levels in the different cells through an unknown mechanism (question mark) might be important for apoptosis induction.
P E R S P E C T I V E S
NATURE REVIEWS | CANCER VOLUME 8 | FEBRUARY 2008 | 143
4. eliminating defective cells by cell competition

Figure 3.1 Examples of programmed cell death (PCD) in plant development and inresponse to environmental fluctuations. PCD occurs during embryogenesis (suspensorelimination), tapetum degeneration, pollen self-incompatibility, organ senescence(petals, sepals, leaves), formation of lace leaf shape, synergid and antipode cell death inthe female gametophyte, tracheary element differentiation, some types of trichomematuration, cell death of the aleurone layer in germinatingmonocot seeds, aerenchymaformationunderoxygen deprivation (anoxia), cell deathof root cap cells, cell death dur-ing allelopathic plant^plant interactions, and plant attack by necrotrophic or hypersen-sitive response (HR)-triggering pathogens.
Gadjev, I (2008) InternaBonal Review of Cell and Molecular Biology, Volume 270
PCD in plants
Pannell, R. and Lamb, C. (1997) The plant cell. 9: 1157-‐1168
• Associate with developmental processes • developmental processes
• embryo formaBon • degeneraBon of the aleurone layer • differenBaBon of tracheary
elements in water-‐conducBng xylem Bssues
• formaBon of root aerenchyma and epidermal trichomes
• anther tapetum degeneraBon • floral organ abscission • pollen self-‐incompaBbility • leaf shape and leaf senescence
• Plant immune response • Can be induced by ROS and H2O2 • VacuolizaBon, nuclear fragmentaBon,
internucleosomal cleavage, and acBvaBon of serine and cysteine proteases

1158 The Plant Cell
aleurone (monocots)
suspensor trichomes
pericarp(unpollinated ovules)
systemic PCD(disease resistance)
HR (disease resistance)
parenchyma (hypoxia)
root cap
Figure 1. Sites of PCD in a Vascular Plant.
The orange spheres represent internal dead cells, and the branched structures on the leaves represent trichomes.
(Figure 1; Nooden, 1988; see also Bleecker and Patterson,1997, in this issue). During interactions with the environment,cell death occurs in the so-called hypersensitive response(HR) to pathogen attack (Figure 1; Lamb and Dixon, 1997).
There is clear evidence that cell death during plant devel-opment and interactions with the environment involves PCD.Table 1 shows some of this evidence, whereas Table 2shows the cells and tissues in which PCD in plants isthought to occur. In this review, we discuss the evidence forPCD during plant vegetative development. PCD is also likelyto occur during the reproductive phase of plant develop-ment, for example, in the deletion of nonfunctional mega-spores and other cells in the germ cell lines (Bell, 1996a).Although the relationship between the mechanisms underly-
ing PCD in animals and plants is not known, we also con-sider the possible evolutionary origins of PCD in plants.
DEVELOPMENT
Aleurone Cells
In seeds of monocots, aleurone cells form a secretory tissuethat releases hydrolases to digest the endosperm and nourishthe embryo. Aleurone cells are unnecessary for postembryonicdevelopment and die as soon as germination is complete(Kuo et al., 1996). Several lines of physiological evidence
Pannell, R. and Lamb, C. (1997) The plant cell. 9: 1157-‐1168
PCD in plants

Biochemical modification of apoptosis
• protein cleavage – By caspase cascade
• protein cross-linking – expression and activation of tissue transglutaminase
• DNA breakdown – Ca2+-and Mg2+-dependent endonucleases
• phagocytic recognition – movement of the normal inward-facing
phosphatidylserine of the cell’s lipid bilayer to expression on the outer layers of the plasma membrane
– presence of annexin I and calreticulin

Process of Apoptosis
• controlled by a diverse range of cell signals – Extrinsic inducers: toxins, hormones, growth
factors, nitric oxide or cytokines – Intrinsic inducers: glucocorticoids, heat,
radiation (DNA damage), nutrient deprivation, viral infection, hypoxia and increased intracellular calcium concentration

Caspases
• have proteolyBc acBvity and are able to cleave proteins at asparBc acid residues
• different caspases have different specificiBes involving recogniBon of neighboring amino acids.
• ten major caspases have been idenBfied – iniBators (caspase-‐2,-‐8,-‐9,-‐10) – effectors or execuBoners (caspase-‐3,-‐6,-‐7) – Inflammatory caspases (caspase-‐1,-‐4,-‐5)
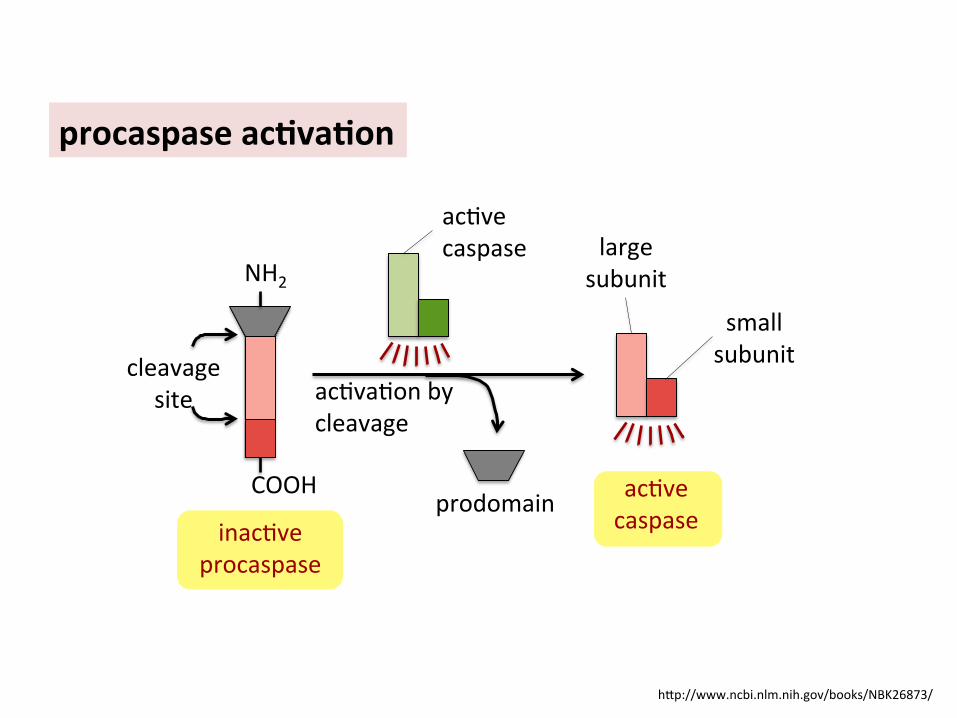
h]p://www.ncbi.nlm.nih.gov/books/NBK26873/
procaspase ac6va6on
acBve caspase
prodomain
acBvaBon by cleavage
acBve caspase large
subunit
small subunit cleavage
site
NH2
COOH
inacBve procaspase

h]p://www.ncbi.nlm.nih.gov/books/NBK26873/
caspase cascade

3 steps of apoptosis
• Initiation (intrinsic and extrinsic) – innitiate by adaptor proteins = initiator procaspases – the initiator procaspases have a small amount of protease
activity – conformation change -> activated and aggregate other
procaspase proteins – the top of the cascade cleaves downstream procaspases to
amplify the death signal and spread it throughout the cell
• Execution – Execution caspases activate cytoplasmic endonuclease and
proteases -> degrade DNA and cytoskeletal proteins
• Phagocytosis – by macrophages, parenchymal cells, or neoplastic cells and degraded within phagolysosomes
Click here to see video link of apoptosis
h]p://bit.ly/1IEo6oi

Intrinsic pathway
• DNA damage, or absence of growth factor -> ATP -> activate p53
• p53 -> activate BAX • BAX make pores at the outermembrane of
mitochondria -> ions kept in intermembrane leak -> cytC
• cytC binds to APAF -> apoptosome • Apoptosome -> activate procaspase 9 ->
caspase cascade
Video = h]p://bit.ly/25FuI38

www.bioscience.org
• a large quaternary protein structure formed in the process of apoptosis • activate the inactive pro-caspase-9 -> apoptosis
Apoptosome

Extrinsic pathway
• Activated by extracellular signals, e.g. TNF (tumor necrosis factor) secreated by natural killer cell
• TNF binds to the TNF receptor (TNFR) at plasma membrane of the cell -> activate dead domain at the cytoplasmic region of the TNFR
• TRADD (TNF receptor associated dead domain) binds to the TNFR
• FADD (Fas associated dead domain) bind to TRADD • Procaspase 8 binds to FADD -> active caspase 8 ->
caspase cascade

Extrinsic pathway
• Activated by extracellular signals, e.g. TNF (tumor necrosis factor) or Fas ligand secreated by natural killer cell
• TNF binds to the TNF receptor (TNFR) or Fas receptor at plasma membrane of the cell -> activate its domain at the cytoplasmic region (so called TRADD or FADD) at the cytoplasmic region
• FADD adapter protein which has DED (Death effector domain) binds to the FADD (dimerization)
• Procaspase 8 binds to DED of FADD adater protein -> active caspase 8 -> caspase cascade
Video = h]p://bit.ly/1RTvy5c

Fas ligand (from lyphocyte)
Fas receptor
FADD (Fas Associated Death Domain)
FADD Adapter protein
Death Effector Domain (DED)
Procaspase 8
Dead inducin
g sig
naling complex
Extrinsic pathway
Picture drew from video of Ben Garside

h]p://www.ncbi.nlm.nih.gov/books/NBK26873/

Elmore, S. (2007) Toxicologic Pathology, 35:495–516,

Genes/proteins inhibiting apoptosis
• Bcl-2 – Inhibit the release of cytC from mitochondria – inhibit Bax-Bax homo dimers forming, which stimulate
apoptosis (p53 is an activator of apoptosis by decreasing Bcl-2 production)
• IAP (inhibitor of apoptosis) – bind to some procaspases to prevent their activation – bind to caspases to inhibit their activity – IAP proteins were discovered as insect viral proteins used to
prevent the infected cell from killing itself before the virus has had time to replicate.
– When mitochondria release cytochrome c to activate Apaf-1, they also release a protein that blocks IAPs, thereby greatly increasing the efficiency of the death activation process.

Bcl-‐2 Bax Bax
Bax Bax
Bax
Bax Bcl-‐2
Bcl-‐2
Bcl-‐2
Bax
Bax
Bax

• p53 – normally inactive (targeted by mdm2 protein) unless
DNA damage, check point proteins will phosphorelate p53
– Phosphorelated p53 will no longer bind to mdm2 and activated
– Activated p53 form tetramer and bind to the promoter of the following genes à increased transcription
• DNA repair genes • p21 (arrests cell cycle) • Pro-apoptotic genes (e.g. Puma, Noxa which binds to antiapoptotic
Bcl2 proteins) à Bax-Bax homodimer à CytC released to cytoplasm à CytC bind to APAF àprocaspase 9 à apoptosis
Genes/proteins inhibiting apoptosis

Apoptotic assays
1. Cytomorphological alteraBons -‐ evaluaBon of hematoxylin and eosin-‐stained Bssue
2. DNA fragmentaBon 3. DetecBon of caspases, cleaved substrates, regulators and inhibitors 4. Membrane alteraBons 5. DetecBon of apoptosis in whole mounts 6. Mitochondrial assays Click here to see video
link of apoptosis h]p://bit.ly/1HJSgqb


Human diseases caused by apoptosis defects
• Immune system dysfunction – Cancer – suppress apoptosis – AIDS – HIV ↑expression of the Fas receptor, resulting in
excessive apoptosis of T cells – Autoimmune lymphoproliferative syndrome (ALPS) – insufficient
apoptosis to destroy aggressive T-cell. Overproduction of B-cell also cause autoimmune
• Neurogenerative diseases – Alzheimer’s – Huntington’s – Parkinson’s – Multiple sclerosis
• Heart disease – ischemia /acute strokes (apoptosis of cadiomycytes)

Autophagy • caspase-independent • major cellular mechanism for eliminate proteins and cytoplasmic
organelles • characterized by
– autophagosomic-lysosomal degradation of bulk cytoplasmic contents – abnormal protein aggregates – excess or damaged organelles
• Require ubiquitin-like protein conjugation system and a protein complex that directs membrane docking and fusion at the lysosome or vacuole
• activated by nutrient deprivation • associated with physiological & pathological processes
– Development – Differentiation – neurodegenerative diseases – Stress (physiology) – Infection – cancer

Importance of autophagy
• plays a role in removing misfolded or aggregated proteins, clearing damaged organelles, such as mitochondria, endoplasmic reBculum and peroxisomes and eliminaBng intracellular pathogens
• promotes cellular senescence • cell surface anBgen presentaBon • protects against genome instability • prevents necrosis • eliminaBng ‘toxic assets’ and promoBng cell viability • prevents diseases:
– Cancer – NeurodegeneraBon – Cardiomyopathy – Diabetes – liver disease – autoimmune diseases – infecBons

www.tanpaku.org

h]p://www.nature.com/ncb/journal/v9/n10/images/ncb1007-‐1102-‐f1.jpg
• Induction • formation of the
autophagosome • fusion with the
lysosome or vacuole • autophagic body
breakdown and recycling.
4 steps of autophagy

h]p://www.wormbook.org/chapters/www_autophagy/autophagyfig1leg.jpg
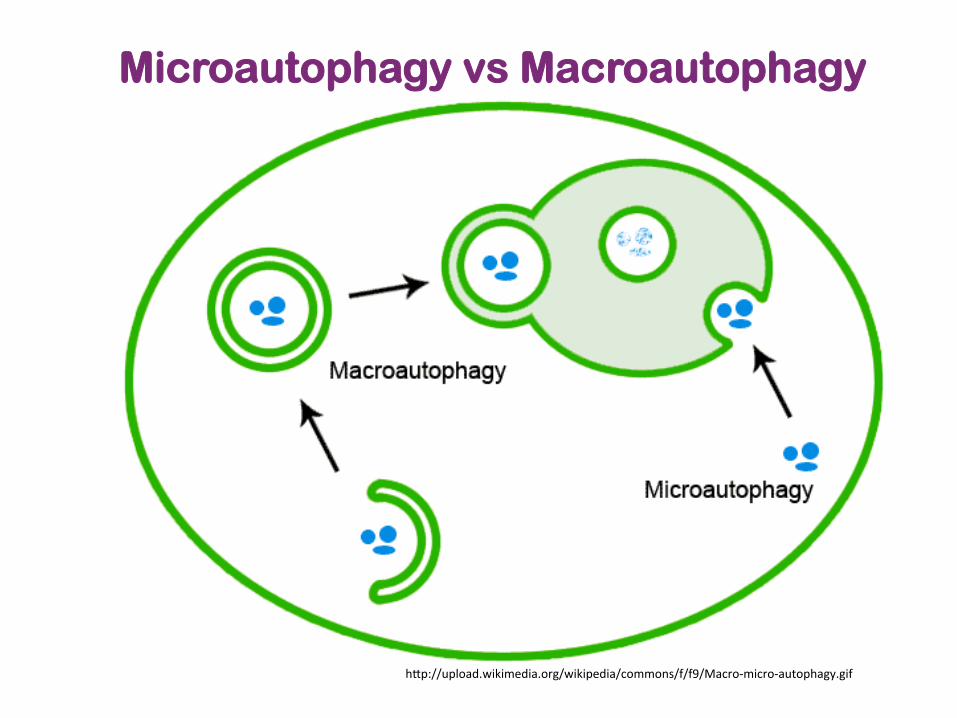
Microautophagy vs Macroautophagy
h]p://upload.wikimedia.org/wikipedia/commons/f/f9/Macro-‐micro-‐autophagy.gif

3 processes of autophagy • Macroautophagy
– main pathway, occurring mainly to eliminate damaged cell organelles or unused proteins.
– formation of autophagosome. – Within the lysosome, the contents of the autophagosome are degraded
via acidic lysosomal hydrolases • Microautophagy
– involves the direct engulfment of cytoplasmic material into the lysosome – occurs by invagination
• Chaperone-mediated autophagy (CMA) – a very complex and specific pathway – involved in the recognition by the hsc70-containing complex – the targeted proteins are recognized by hsc70 complex -> bind to
chaperone -> forming the CMA- substrate/chaperone complex -> moves to the lysosomal membrane-bound protein and enter the cell via CMA receptor
– the substrate protein gets unfolded and it is translocated across the lysosome membrane with the assistance of the lysosomal hsc70 chaperone.
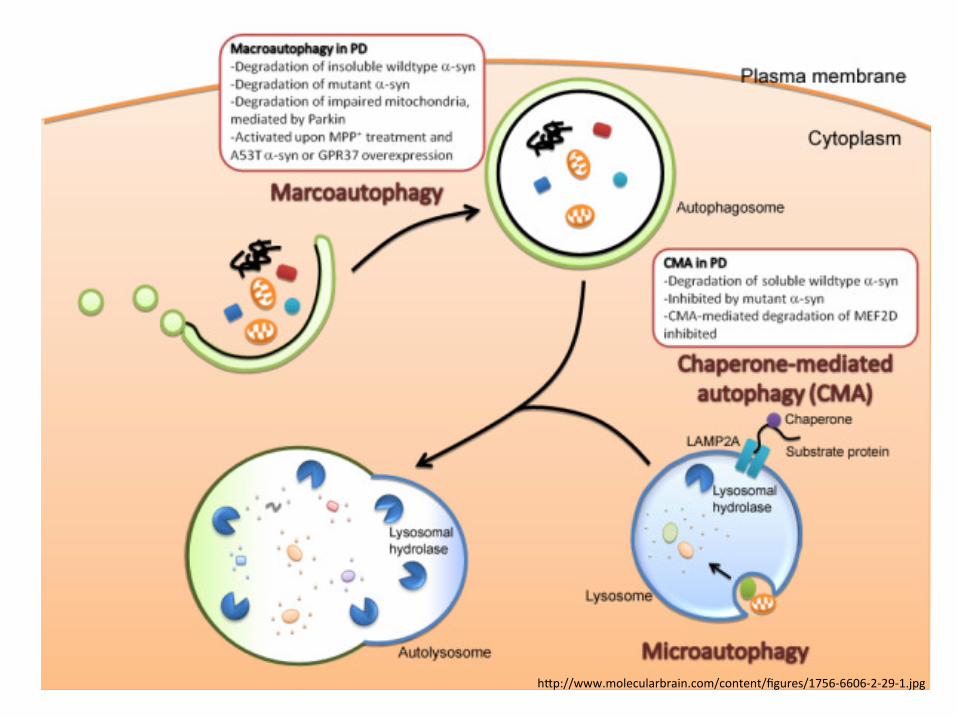
h]p://www.molecularbrain.com/content/figures/1756-‐6606-‐2-‐29-‐1.jpg


Autophagy vs. apoptosis
• show some ultrastructural features characterisBc of apoptosis – mitochondria may be central organelles that integrate both apoptosis and autophagy
– Used same signals e.g. Akt (protein kinase B) and p70S6 kinase, DAPk, Beclin 1, BNIP3, HSpin1, or protymosin-‐α

References • http://www.ncbi.nlm.nih.gov/books/NBK26873/ • http://www.discoverymedicine.com/Jie-Shen/2009/11/24/programmed-cell-death-and-apoptosis-in-aging-and-life-
span-regulation/ • http://www.enzolifesciences.com/browse/cell-death-apoptosis-autophagy/ • http://www.youtube.com/watch?v=xdLPpdoU2Nc • http://www.youtube.com/watch?v=wREkXDiTkPs • Li, F., Huang, Q., Chen, J., Peng, Y., Roop, D. R., Bedford, J. S., and Li, C.-Y. (2010). Apoptotic cells activate the
“phoenix rising” pathway to promote wound healing and tissue regeneration. Sci. Signal. 3, ra13. • Gadjev, I., Stone, J. M., and Gechev, T. S. (2008). Programmed cell death in plants: new insights into redox
regulation and the role of hydrogen peroxide. Int. Rev. Cell Mol. Biol. 270, 87–144. • Reape, T. J., Molony, E. M., and McCabe, P. F. (2008). Programmed cell death in plants: distinguishing between
different modes. J. Exp. Bot. 59, 435–44. • Xie, Z., and Klionsky, D. J. (2007). Autophagosome formation : core machinery and adaptations. Nat. Cell Biol. 9,
7–14. • Glick, D., Barth, S., and Macleod, K. F. (2010). Autophagy : cellular and molecular mechanisms. J. Pathol. 221, 3–
12. • Elmore, S. (2007). Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516. • Cova, C. De, Abril, M., Bellosta, P., Gallant, P., and Johnston, L. A. (2004). Drosophila Myc Regulates Organ Size
by Inducing Cell Competition. 117, 107–116. • Greenhalgh, D. G. (1998). The role of apoptosis in wound healing. Int. J. Biochem. cell Biol. 30, 1019–1030. • Havel, L., and Durzan, D. J. (1996). apoptosis in plant. Bot. Acta 109, 268–277. • Fuchs, Y., and Steller, H. (2011). Programmed cell death in animal development and disease. Cell 147, 742–58. • Shen, J., and Tower, J. (2009). Programmed Cell Death and Apoptosis in Aging and Life Span Regulation.
Discov. Med. 8, 223–226. • Reynaud, K., and Driancourt, M. a (2000). Oocyte attrition. Mol. Cell. Endocrinol. 163, 101–8. • Greenberg, J. T. (1996). Review Programmed cell death : A way of life for plants. Proc. Natl. Acad. Sci. U. S. A.
93, 12094–12097.