a meta-analysis of the microbial diversity observed in anaerobic digesters
TRANSCRIPT

Bioresource Technology 102 (2011) 3730–3739
Contents lists available at ScienceDirect
Bioresource Technology
journal homepage: www.elsevier .com/locate /bior tech
A meta-analysis of the microbial diversity observed in anaerobic digesters
Michael C. Nelson a, Mark Morrison a,b, Zhongtang Yu a,b,⇑a Environmental Science Graduate Program, The Ohio State University, Columbus, OH 43210, USAb Department of Animal Sciences, The Ohio State University, Columbus, OH 43210, USA
a r t i c l e i n f o a b s t r a c t
Article history:Received 31 August 2010Received in revised form 23 November 2010Accepted 25 November 2010Available online 3 December 2010
Keywords:Anaerobic digestionMethaneBiogasBiomethanationMicrobial diversity
0960-8524/$ - see front matter � 2010 Elsevier Ltd. Adoi:10.1016/j.biortech.2010.11.119
⇑ Corresponding author at: Department of AnimUniversity, Columbus, OH 43210, USA. Tel.: +1 614 292
E-mail address: [email protected] (Z. Yu).
In this study, the collective microbial diversity in anaerobic digesters was examined using a meta-anal-ysis approach. All 16S rRNA gene sequences recovered from anaerobic digesters available in public dat-abases were retrieved and subjected to phylogenetic and statistical analyses. As of May 2010, 16,519bacterial and 2869 archaeal sequences were found in GenBank. The bacterial sequences were assignedto 5926 operational taxonomic units (OTUs, based on P97% sequence identity) representing 28 knownbacterial phyla, with Proteobacteria (1590 OTUs), Firmicutes (1352 OTUs), Bacteroidetes (705 OTUs), andChloroflexi (693 OTUs) being predominant. Archaeal sequences were assigned to 296 OTUs, primarilyMethanosaeta and the uncharacterized WSA2 group. Nearly 60% of all sequences could not be classifiedto any established genus. Rarefaction analysis indicates that approximately 60% of bacterial and 90% ofarchaeal diversity in anaerobic digesters has been sampled. This analysis of the global bacterial andarchaeal diversity in AD systems can guide future studies to further examine the microbial diversityinvolved in AD and development of comprehensive analytical tools.
� 2010 Elsevier Ltd. All rights reserved.
1. Introduction
Anaerobic digestion (AD) of organic wastes is a microbiallymediated process whereby complex organic wastes are ultimatelyconverted into methane biogas, a potential renewable energysource. The overall AD process can be conceptually divided intofour phases defined by the primary catabolic reactions that occurat each phase: hydrolysis of complex polymers (I, hydrolysis),fermentation of the hydrolysis end-products to short chain fattyacids (SCFAs) (II, acidogenesis), conversion of SCFAs of three ormore carbons to primarily acetate (III, syntrophic acetogenesis),and the production of methane (IV, methanogenesis) (Yu et al.,2010). The guilds of microbes involved in each phase of AD areinterdependent through cross-feeding and/or maintenance ofchemothermodynamic gradients. As a result the AD process canquickly and easily breakdown when one of the four phases is outof balance, such as an accumulation of SCFAs that can lead to acid-ification of the entire system (Chen et al., 2008). Failures of the ADprocess in bioreactors treating high strength organic wastes fromindustrial or agricultural operations can lead to damagingeconomic losses. As AD is increasingly looked upon as a source ofbioenergy, the reliability and stability of AD systems becomescritical to ensuring both reliable energy supplies and uninterruptedcore business operations.
ll rights reserved.
al Sciences, The Ohio State3057; fax: +1 614 292 2929.
Numerous studies have been conducted to gain a better under-standing of the microbiomes present in AD reactors and theirinfluence on the efficiency and stability of the AD processes (e.g.reviewed in Chen et al., 2008). While initial studies employedtraditional cultivation-based methods, the primary methods incurrent use are DNA-based molecular biology methods such ascloning and sequencing of either functional or 16S ribosomalRNA (rRNA) genes, FISH, DGGE, single-stranded conformation poly-morphisms (SSCP), and quantitative PCR (Leclerc et al., 2004; Malinand Illmer, 2008; Sousa et al., 2007). Because it allows for identifi-cation of potential known and unknown microbes present in ADreactors, cloning and sequencing of 16S rRNA genes has beengenerally favored over other methods.
Most studies to date, however, have focused on a single specificAD system (e.g. upflow anaerobic sludge bed (UASB) reactors orcontinuous stirred tank reactors (CSTRs) processing a single wastestream (e.g. municipal sewage, brewery wastewater). Many of thedatasets published contain a small numbers of cloned sequences(generally <100), thus revealing only a small portion of the fulldiversity present in anaerobic digesters (e.g. Lefebvre et al.,2007). Some of these studies were further limited by a narrowfocus on one particular microbial group such as the Archaea or aparticular phylum (e.g. Chouari et al., 2003; Hori et al., 2006). Addi-tionally, many sequences recovered from AD systems were depos-ited into GenBank but have not yet been reported in the literature,contributing little to no additional information on the microbialdiversity and its function. As a result, the understanding of themicrobiomes involved in AD is fragmented and likely biased,

M.C. Nelson et al. / Bioresource Technology 102 (2011) 3730–3739 3731
exemplified by these microbiomes still being regarded as a ‘‘blackbox’’. This knowledge gap limits the understanding of how thesecomplex microbiomes either hamper or enhance the efficiencyand stability of AD systems.
A few studies have examined the microbial diversity of anaero-bic digesters using relatively large (>200 sequences) 16S rRNAclone libraries (Chouari et al., 2005; Figuerola and Erijman, 2007;Godon et al., 1997; Rivière et al., 2009). Additionally, a few studieshave used 454-pyrosequencing, either alone or in combinationwith the Sanger sequencing technology, to analyze the microbio-mes in anaerobic digesters, producing large datasets of short, diffi-cult to classify sequence reads (Krause et al., 2008; Kröber et al.,2009; Schlüter et al., 2008; Zhang et al., 2009). To date, however,there has been no collective overview of the microbial diversitygenerally found in AD systems. In this study, a meta-analysis wasperformed on all publicly available 16S rRNA gene sequences gen-erated by Sanger sequencing from various anaerobic digesters inan effort to provide a collective appraisal of the microbial diversityin AD systems. Estimates of the current coverage of the microbialdiversity already identified in anaerobic digesters were made andparticular gaps in the knowledge and understanding of the micro-bial populations involved in AD were identified.
2. Methods
2.1. Sequence data collection
Initial sequence sets were obtained from the GenBank(http://www.ncbi.nlm.nih.gov) and RDP (Release 10, http://rdp.cme.msu.edu) databases using the search terms ‘anaerobic di-gester’, ‘bioreactor’, ‘CSTR’, and ‘UASB’ in the months through May01, 2010. All non-16S rRNA sequences were removed and theresulting composite dataset was de-replicated to remove identicalrecords based on Accession Number. Sequences not recovered frommethanogenic AD systems, particularly those corresponding onlywith heavy metal and chlorinated solvent remediation, were man-ually removed according to the annotation provided in the Gen-Bank sequence records. Published datasets that were notautomatically retrieved using the search terms were manuallyadded. Sequences with vector nucleotides were trimmed to leaveonly nucleotides confirmed as rRNA after alignment against the16S reference sequences from Escherichia coli (Accession Number:U00096) for bacteria or Methanothermobacter thermoautotrophicus(Accession Number: AE000666) for archaea. Sequences shorterthan 250 bp were removed from the dataset to avoid uncertaintiesin comparing and classifying short sequences that have little or nosequence overlap. The remaining sequences comprised the re-dacted composite dataset used in this study.
2.2. Phylogenetic analyses
Sequences were grouped into batches of roughly 5000 se-quences by size such that the shortest sequence was no more than20% shorter than the longest sequence in each batch. Batches were
Table 1Diversity satistics for Archaea, Bacteria, and ‘Major’ phylum groups.
Group Total sequences % Unclassified to phylum # of OTUsa
Archaea 2869 2.15 296Bacteria 16519 16.28 5926Chloroflexi 3744 – 693Proteobacteria 3585 – 1590Firmicutes 2549 – 1352Bacteroidetes 2436 – 705
a Values were calculated using a 0.03 dissimilarity cut-off.b Coverage = # OTUs / Rarefaction Estimate.
submitted for NAST alignment with the minimum alignmentlength set to 80% of the shortest sequence in each batch and allother criteria using default values (DeSantis et al., 2006). Theresulting aligned sequences were imported into ARB and insertedinto the Greengenes database ARB tree using the positional vari-ance by parsimony method (Ludwig et al., 2004). Unaligned se-quences were classified en masse to taxonomic ranks with theClassifier program implemented as part of the RDP database usingdefault parameters (Wang et al., 2007). Based on the classificationsdetermined with the Classifier program, distance matrices werecomputed within ARB using Jukes-Cantor correction for the follow-ing groups: Archaea, Bacteria, Proteobacteria, Firmicutes, Bacteroide-tes, Chloroflexi, and the collected ‘‘minor phyla’’ of Bacteria thatcomprised sequences not assigned to any of the aforementionedphyla. Individual distance matrices were analyzed using MOTHURto cluster OTUs, generate rarefaction curves, and determine thenonparametric ACE and Chao1 richness estimates (Schloss et al.,2009). A parametric estimation of expected maximum number ofOTUs was conducted using the non-linear models procedure (PROCNLIN) of SAS (V9.1, SAS Inst. Inc., Cary, NC). This method fits themonomolecular function to the rarefaction output generated byMOTHUR to determine the asymptote that serves as the upperbound of the curves as previously described (Larue et al., 2005).The value defined by the asymptote is an estimate of the expectedmaximum species richness complementary to the ACE and Chao1estimates and has been used previously to estimate maximum spe-cies richness (Larue et al., 2005; Youssef and Elshahed, 2008). Un-less otherwise stated, the term OTU was defined as a grouping ofsequences that share 60.03 sequence dissimilarity and is takento represent the species taxonomic rank. The following dissimilar-ity cut-offs were used to approximate other taxonomic ranks: 0.05,genus; 0.10, family; 0.15, class/order; 0.20, phylum (Schloss andHandelsman, 2004). A treemap based on the output from theRDP Classifier was constructed using version 4.1.1 of the programTreemap (http://www.cs.umd.edu/hcil/treemap).
2.3. Nucleotide Accession Numbers
The Accession Numbers for all sequences analyzed in this studyare available from the corresponding author. The sequences arecurrently maintained in an in-house ARB database of anaerobic di-gester sequences. A copy of this database and the sequence align-ment are also available by request from the corresponding author.
3. Results and discussion
This study was conducted as a naïve meta-analysis of allpublicly available 16S rRNA gene sequences recovered from ADreactors worldwide. The term naïve is used here to imply thatsequences were collected and analyzed irrespective of theirpreviously determined taxonomic associations or other analyses.As AD becomes an increasingly engineered process for wastemanagement and biogas production, it becomes necessaryto understand the totality of microorganisms that are able to
ACEa Chao1a Rarefaction estimationa Current coverageb (%)
362 336 327 9020538 11717 9646 61
3238 1858 1157 606548 3498 2658 603184 2674 2298 591494 1221 1076 66

Fig. 1. Phylogenetic tree of OTU sequences. Phylogenetic tree of OTU sequences with phylum level branches grouped and labeled. Individual sequences, where shown, arelabeled with their Accession Number.
3732 M.C. Nelson et al. / Bioresource Technology 102 (2011) 3730–3739
participate in the process. Thus, the results represent a fresh, up-dated, and global view of the microbial diversity involved in theAD process in general.
3.1. Data summary
A total of 19,388 sequences (16,519 bacterial and 2869 archa-eal) were retrieved and analyzed. The bacterial sequences wereassigned to 5935 OTUs, while the archaeal sequences were as-
signed to 296 OTUs. The most abundant bacterial OTU contained1799 sequences while the most abundant archaeal OTU con-tained 480 sequences. Of the bacterial sequences analyzed, amajority (approx. 63%) were classified within four ‘major’ phyla:Chloroflexi, Proteobacteria, Firmicutes, and Bacteroidetes (Table 1and Figs. 1 and 2). Based on assignment using the RDP Classifier,1667 of the bacterial sequences were listed as ‘‘unclassified’’ atthe phylum level. The remaining sequences were classifiedwithin 24 ‘minor’ phyla (Fig. S2), of which the Synergistetes,
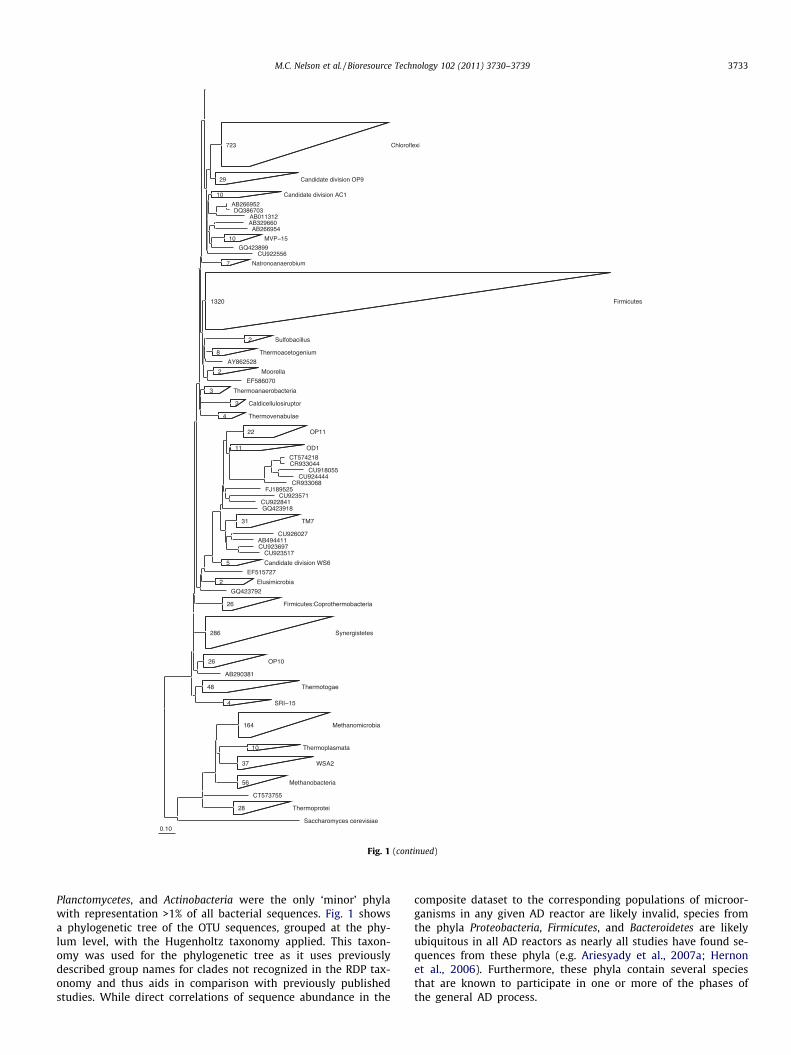
Fig. 1 (continued)
M.C. Nelson et al. / Bioresource Technology 102 (2011) 3730–3739 3733
Planctomycetes, and Actinobacteria were the only ‘minor’ phylawith representation >1% of all bacterial sequences. Fig. 1 showsa phylogenetic tree of the OTU sequences, grouped at the phy-lum level, with the Hugenholtz taxonomy applied. This taxon-omy was used for the phylogenetic tree as it uses previouslydescribed group names for clades not recognized in the RDP tax-onomy and thus aids in comparison with previously publishedstudies. While direct correlations of sequence abundance in the
composite dataset to the corresponding populations of microor-ganisms in any given AD reactor are likely invalid, species fromthe phyla Proteobacteria, Firmicutes, and Bacteroidetes are likelyubiquitous in all AD reactors as nearly all studies have found se-quences from these phyla (e.g. Ariesyady et al., 2007a; Hernonet al., 2006). Furthermore, these phyla contain several speciesthat are known to participate in one or more of the phases ofthe general AD process.

3734 M.C. Nelson et al. / Bioresource Technology 102 (2011) 3730–3739
3.2. Archaea
The majority of the archaeal sequences, nearly 95%, were classi-fied within the phylum Euryarchaeota, with only 139 sequencesbeing classified within Crenarchaeota and 22 sequences failing tobe classified to any of the recognized archaeal phyla. The crenar-chaeal sequences were confined solely to the class Thermoprotei.Among the Euryarchaeota, 888 sequences were not assigned toany existing class, with the remaining sequences being assignedprimarily to the methanogenic classes Methanomicrobia (1590sequences) and Methanobacteria (223 sequences), with sevensequences assigned to the genus Thermogymnomonas in theclass Thermoplasmata. The small numbers of Crenarchaeota andThermoplasmata sequences suggests that these two groups areeither allochthonous to or scarce in AD reactors and are probablynot functionally vital in the AD process.
As seen in Fig. 2, the obligately acetoclastic Methanosaeta (for-merly Methanothrix) was the most predominant archaeal genus,representing approximately 55% of the archaeal sequences as-signed to a genus. Methanospirillum, which preferentially utilizesH2/CO2 over formate as substrate, and the obligately hydrogeno-trophic genus Methanobacterium each represented nearly 10% ofthe archaeal sequences assigned to a genus. The genera Methano-culleus, Methanolinea, and Methanosarcina each representedroughly 5% of genus-assigned sequences. The other 10 methanogengenera were only represented by a small number of sequences inthe dataset (Fig. S1). These results are in general agreement withthe finding reported in the literature that the predominant aceto-clastic methanogens in AD systems are Methanosaeta spp. (Leclercet al., 2004).
While the RDP taxonomic assignments, as shown in Fig. 2, sug-gest a large number of unclassified Euryarchaeota, the phylogenetictree based on the Hugenholtz taxonomy (Fig. 1) showed that mostof these sequences belonged to the uncultured WSA2 group ofmethanogens. This WSA2 group, also known as ArcI, has beenfound in several AD systems and currently has no cultured repre-sentatives (Chouari et al., 2005; Rivière et al., 2009). The lack ofcultured isolates with high similarity to this group of sequencesmakes it difficult to infer the function (e.g. substrates) of the meth-anogens represented by these sequences, however work by Chou-ari et al. (2005) suggested that members of this group representsmethanogens are capable of growth at least on H2/CO2.
3.3. Bacteria
3.3.1. ChloroflexiThe Chloroflexi comprised a total of 3744 sequences, approxi-
mately 22% of the bacterial sequences. Compared to the other three‘major phyla’, however, Chloroflexi diversity was the lowest withonly 693 OTUs generated and a Simpson’s Index of 0.14, which ismuch greater than that for the other three major phyla. Highlight-ing the unevenness of the phylum, over 60% of all the Chloroflexi se-quences (13% of all bacterial sequences) were assigned to six OTUsclassified as ‘‘unclassified Anaerolineaceae’’. Overall, just over 90%of the Chloroflexi sequences were assigned to unclassified Anaero-lineaceae, while less than 5% were assigned to a known genus(Fig. 2). The eight genera that were identified include all five gen-era in the family Anaerolineaceae, Caldilinea, Dehalogenimonas, andSphaerobacter (Fig. S2). The majority of all the Chloroflexi sequenceswere found by a group of researchers in France through multiplestudies investigating the microbial diversity in digested sludgefrom municipal wastewater treatment plants located in France,Germany, and Chile (Chouari et al., 2005; Rivière et al., 2009).Although it is not possible to infer their functions in anaerobicdigesters, the bacteria represented by these OTUs could play a sig-nificant role, at least, in the municipal sludge digesters where these
sequences were recovered. Future studies are needed to examinethe distribution and abundance of Chloroflexi species in other typesof anaerobic digesters.
3.3.2. ProteobacteriaThe Proteobacteria was the second largest and yet most diverse
phylum in the dataset. While 159 fewer sequences were assignedto the Proteobacteria (3585) than the Chloroflexi, there were morethan twice as many OTUs identified (1590), with a Simpson’s Indexnearly two orders of magnitude lower. Unlike the Chloroflexisequences, the majority (approx. 70%) of the proteobacterialsequences were assigned to a total of 169 known genera(Fig. S3). All five classes within the Proteobacteria were repre-sented, but the Beta-, Alpha-, and Deltaproteobacteria togetherrepresented roughly 86% of the proteobacterial sequences. TheEpsilonproteobacteria represented only 58 sequences, withsequences classified to the genus Arcobacter comprising 28 ofthose. The Gammaproteobacteria represented 378 sequences, withthe genus Pseudomonas representing 17% of those. Only 43sequences, approximately 0.25% of all bacterial sequences, wereclassified within Enterobacteriales, indicating a low recovery ratefrom most AD reactors of enteric bacteria that include manycommon human enteric pathogens found in sewage.
The fourth largest bacterial OTU in the dataset was assigned tothe genus Brachymonas, which was the most numerically repre-sented genus in the Proteobacteria. This genus is capable of denitri-fication, and along with other members of the Comamonadaceae, isable to degrade organic acids including acetate. Some members ofthis OTU may be responsible for denitrification in some anaerobicdigesters, such as utilizing the nitrate that is produced in the aer-obic treatment process of wastewater treatment plants. The generaRhodobacter and Thauera are similarly capable of growth on organicacids and were the second and fourth most abundant proteobacte-rial genera, respectively. The third most abundant genus, Smithella,is known to participate in the syntrophic oxidation of propionate toacetate for use by acetoclastic methanogens (Ariesyady et al.,2007b). As propionate is an important fermentation product fromphase II (Speece et al., 2006), it is not surprising that syntrophicpropionate consumers such as Smithella and Syntrophobacter werecorrespondingly represented by a large number of sequences in thedataset.
3.3.3. FirmicutesThe third largest identified phylum was the Firmicutes, with
2549 sequences assigned. A majority of the sequences within theFirmicutes, nearly 86%, were assigned to the class Clostridia. Theclass Bacilli was represented by only 172 sequences, while the classErysipelotrichi was represented by only 42 sequences. ‘UnclassifiedFirmicutes’ comprised 150 sequences. A total of 108 recognizedgenera were identified, comprising 1141 sequences (Fig. S4). With-in the class Clostridia, over 1/3 of the sequences were either unclas-sified to any known order or were unclassified below the familyClostridiales (Fig. 2). A further 6.7% were determined to be‘Unclassified Ruminococcaceae’, a family that contains a numberof cellulolytic and amylolytic species isolated from several typesof anaerobic environments, including AD reactors.
All genera comprising greater than 5% of Firmicutes sequenceswere found within the class Clostridia (Fig. S4). The cellulolyticgenus Acetivibrio was the predominant genus with 162 assignedsequences, while the genus Clostridium was second in abundancewith 116 sequences. Both of these genera include of a number ofspecies that are known to degrade complex biopolymers such ascellulose. This observation corroborates the finding that celluloseis a component of many AD feedstocks and that cellulolyticbacteria are important participants in phase I of the AD process(Burrell et al., 2004; Yu et al., 2010). The proteolytic genera

Fig. 2. Treemap of observed taxons shown in their hierarchical order. Treemap showing taxonomic ranking of all taxa with greater than 50 sequences for all 19,388 retrievedsequences. Taxonomic ranks are shown hierarchically as boxes within boxes, with each rank color coded as follows: Domain – white, Phylum – red, Class – orange, Order –yellow, Family – green, Genus - blue. The size of each box is proportional to the number of sequences assigned to that taxon with respect to the entire dataset. Each box islabeled according to its RDP classification, with domain and bacterial phylum headings and end-rank boxes including the number of sequences represented if size permits.The placement of boxes is arbitrary with respect to boxes within the same taxonomic rank and does not correspond to any form of phylogeny or relatedness. aUnclassifiedLachnospiraceae. bUnclassified Veillonellaceae.
M.C. Nelson et al. / Bioresource Technology 102 (2011) 3730–3739 3735
Coprothermobacter and Sedimentibacter represented 104 and 103sequences, respectively. The only other genus representing morethan 5% of Firmicutes sequences is Syntrophomonas, a genus com-prised of syntrophic acetogens capable of butyrate and propionatedegradation. Within the class Bacilli, the two primary genera wereLactococcus and Bacillus, representing 31 and 29 sequences,respectively.
3.3.4. BacteroidetesApproximately 50% of the Bacteroidetes sequences, including the
second largest bacterial OTU in the dataset and two other OTUsrepresenting more than 50 sequences, were unable to be classifiedto any known class, with a further 190 sequences assigned to thegroup ‘unclassified Bacteroidales’ (Fig. 2). In total, only about 30%of the Bacteroidetes sequences were able to be classified to one of30 recognized genera (Fig. S2). The ninth largest OTU within thebacterial dataset was assigned to the most abundant Bacteroidetesgenus, Paludibacter, which in total represented 217 sequences.Paludibacter is exemplified by the type strain P. propionicigenesWB4, a mesophilic anaerobic bacterium that produces propionateand acetate as its primary fermentation products (Ueki et al.,2006). The second largest genus in this phylum was Prolixibacter,which is typified by P. bellariivorans F2, a psychrotolerant organismcapable of sugar fermentation via the mixed acid pathway (Holmeset al., 2007). The third largest genus within the Bacteroidetes isParabacteroides, which is saccharolytic and produces acetate andsuccinate as its primary fermentation end products. Proteiniphilum,the 4th largest genus, is defined by the proteolytic type strainP. acetatigenes initially isolated from a mesophilic UASB (Chenand Dong, 2005).
3.3.5. Minor phylaIn addition to the above discussed ‘major’ phyla, 24 ‘minor’
phyla were represented by the retrieved sequences (Fig. 1,Fig. S2). Of the 24 ‘minor’ phyla, only the Synergistetes, Planctomy-cetes, and Actinobacteria represented more than 1% of all the bac-terial sequences. Some known genera were represented in these‘minor phyla’ (Fig. 2, Fig. S2). The genus Cloacibacillus, withinthe Synergistetes, contained the 5th and 7th largest OTUs in thebacterial dataset and was the single largest identified genusamong the Bacteria. The type strain for this genus, C. evryensisstr. 158, was isolated from an AD reactor at a municipal wastewa-ter treatment plant and is capable of anaerobic protein degrada-tion (Ganesan et al., 2008). The phylum Synergistetes itself wasrecently defined and includes the genus Aminobacterium, to which176 sequences were assigned, as well as other protein-degradingbacteria. Comprising just over 6% of the bacterial sequences inthe dataset, members of the Synergistetes most likely play animportant role in the acidogenic phase (phase II) of the AD pro-cess in AD systems.
The Planctomycetes was the second most abundant of the minorphyla, however the majority of the sequences (60%) were derivedfrom a single study using Planctomycetes-specific primers (Chouariet al., 2003). A further 30% were derived from the large-scalesequencing effort by Rivière et al. (2009), which included the samedigester sampled by Chouari et al. (2003, 2005). Excluding thesesequences gives only 40 sequences from 27 separate studies, sug-gesting a low and/or sporadic occurrence of Planctomycetes in typ-ical AD systems. Within the phylum Actinobacteria, 26% of thesequences were associated with the propionate-producing subor-der Propionibacterinaea, a result that highlights the importance of
a: Archaea
0 1000 2000 30000
50
100
150
200
250
300
3500.030.050.100.20
Num
ber o
f O
TUs
Gen
erat
ed
b: Bacteria
0 2500 5000 7500 10000 12500 15000 17500 0
500100015002000250030003500400045005000550060006500
0.030.050.100.20
Number of Sequences Analyzed
Number of Sequences Analyzed
Number of Sequences Analyzed
Num
ber o
f OTU
s G
ener
ated
c: 'Major' Phyla
0 1000 2000 3000 40000
250
500
750
1000
1250
1500 Chloroflexi
Proteobacteria
Firmicutes
Bacteroidetes
Num
ber o
f OTU
s G
ener
ated
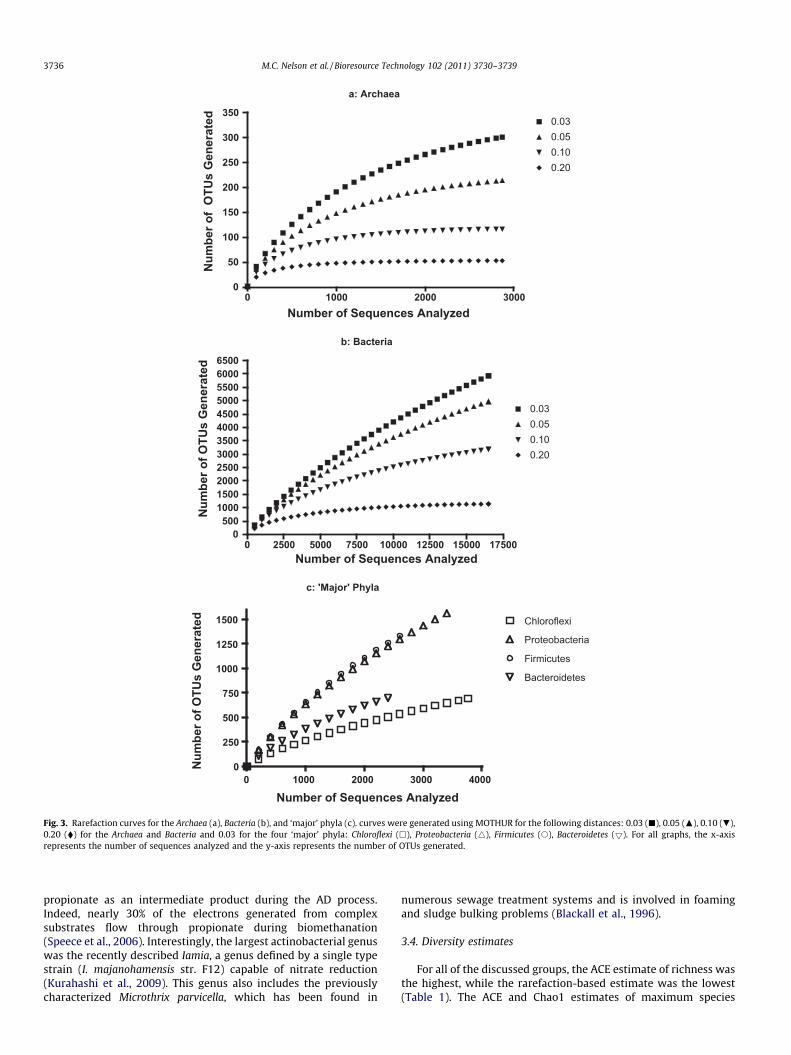
Fig. 3. Rarefaction curves for the Archaea (a), Bacteria (b), and ‘major’ phyla (c). curves were generated using MOTHUR for the following distances: 0.03 (j), 0.05 (N), 0.10 (.),0.20 (�) for the Archaea and Bacteria and 0.03 for the four ‘major’ phyla: Chloroflexi (h), Proteobacteria (4), Firmicutes (s), Bacteroidetes (5). For all graphs, the x-axisrepresents the number of sequences analyzed and the y-axis represents the number of OTUs generated.
3736 M.C. Nelson et al. / Bioresource Technology 102 (2011) 3730–3739
propionate as an intermediate product during the AD process.Indeed, nearly 30% of the electrons generated from complexsubstrates flow through propionate during biomethanation(Speece et al., 2006). Interestingly, the largest actinobacterial genuswas the recently described Iamia, a genus defined by a single typestrain (I. majanohamensis str. F12) capable of nitrate reduction(Kurahashi et al., 2009). This genus also includes the previouslycharacterized Microthrix parvicella, which has been found in
numerous sewage treatment systems and is involved in foamingand sludge bulking problems (Blackall et al., 1996).
3.4. Diversity estimates
For all of the discussed groups, the ACE estimate of richness wasthe highest, while the rarefaction-based estimate was the lowest(Table 1). The ACE and Chao1 estimates of maximum species

M.C. Nelson et al. / Bioresource Technology 102 (2011) 3730–3739 3737
richness differed greatly (43–47%) for the Bacteria, Chloroflexi, andProteobacteria, while the corresponding estimates for theArchaea, Firmicutes, and Bacteroidetes were less than 19% different.These differences stem from the method of correction each calcu-lation uses and have been discussed elsewhere (Hughes et al.,2001). In comparison, the richness estimates derived from the rar-efaction curves differed much less from the Chao1 estimate thanfrom the ACE estimate. Whereas individual studies represent thealpha diversity of microorganisms in a given digester, the compos-ite dataset represents the global diversity of microorganisms in ADsystems in general. Given the large array of options in digester de-sign, operation, feedstock, and geographic region, it should not besurprising that the global diversity seen in the composite dataset ismuch greater than the alpha diversity reported for an individualdigester.
Despite numerous previous studies, the results of this studyshowed that the complete microbial diversity in AD reactors re-mains to be detailed. Rarefaction analysis of the Archaea showedthat sampling at the phylum (0.20 phylogenetic distance) and fam-ily (0.10 phylogenetic distance) levels has reached a maximum,while sampling at the species (0.03 phylogenetic distance) levelis incomplete (Fig. 3). For the Bacteria, only sampling at the phylumlevel has neared its expected maximum while the curves for lowertaxonomic ranks are still projecting upward. This is corroboratedby the estimation of current coverage at the various taxonomiclevels for the Bacteria (Table 2). At the species level, 90% of theexpected archaeal diversity has already been witnessed, while only61% of the bacterial diversity has been revealed. Rarefaction curvesfor the four ‘major’ phyla suggest that sampling of the Chloroflexiand Bacteroidetes is more complete than that of the Proteobacteriaand Firmicutes as the curves have begun to reach a horizontal pla-teau (Fig. 3c). The estimation of current coverage, however, showsthat all four ‘major’ phyla have a similar percent coverage at thespecies level, which is similar to that of the Bacteria (Table 1). Thisis due to there being fewer unobserved OTUs for the Chloroflexi andBacteroidetes than for the Proteobacteria and Firmicutes, eventhough the percent coverage is similar. Rarefaction analysis andthe diversity statistics show that the known bacterial diversity inAD systems is incomplete below the phylum level. Nevertheless,the global diversity revealed in this study may serve as a frame-work for future studies of alpha and beta diversity. More specifi-cally, the collective sequence dataset can be useful in developingmolecular tools such as primers and probes to detect and quantifyspecific groups of either bacteria or archaea. Knowledge arisingfrom such studies will shine light on the ecology of individual bac-teria or archaea found in the dataset.
Current second generation sequencing technologies are capableof providing sufficient coverage and depth to fully explore an indi-vidual sample or compare multiple samples through multiplexing.Studies examining multiple AD reactor designs, being fed differingfeedstocks and operated under various conditions, will not onlygreatly increase the number of sequences available for analysis,
Table 2Estimates of current taxonomic coverage for Archaea and Bacteria.
Distance # OTUs Rarefaction estimation Current coveragea (%)
Archaea0.03 296 327 900.05 209 223 940.1 112 116 970.2 49 52 94Bacteria0.03 5926 9646 610.05 4981 7195 690.1 3186 3754 850.2 1142 1150 99
a Coverage = #OTUs/rarefaction estimate.
but also provide better datasets upon which to perform compari-sons of the beta microbial diversity between individual AD sys-tems. Such knowledge can aide in the design and operation of asystem uniquely tailored to a specific feedstock or application,such as energy production. Additionally, the new sequences canbe added to the composite sequence datasets analyzed in thisstudy to help define the full diversity in anaerobic digesters.Knowledge on the full diversity may greatly advance our under-standing of the microbiome underpinning the AD process and de-fine the significance of individual bacteria and archaea.
One issue that the composite dataset cannot currently addressis the beta diversity of species when comparing two or more ADsystems. While the dataset contained sequences recovered froma wide array of digesters, beta diversity was unable to be deter-mined because few studies generated both large sequence datasetsand provided detailed information (e.g. design, feedstock, opera-tion, etc.) on the AD system analyzed. The different methodologiesand sequence submission criteria used by different researchersalso comprises such analyses. Rivière et al. (2009) recently defineda ‘‘core group alpha’’ of six bacterial OTUs from analyzing sevenmunicipal sludge digesters. This ‘‘core group alpha’’ is comprisedof bacteria affiliated with Bacteroidetes, Chloroflexi, Synergistetesand Betaproteobacteria. While these three phyla or class wereabundant in the composite dataset analyzed in this study,members of the Firmicutes or Alpha- and Delta-Proteobacteria wereabsent in core group alpha. Although it is possible that the uniqueenvironment within a specific AD reactor can select for a distinctmicrobiome, it is intriguing that only a small number of ‘core OTUs’can be found among the large numbers of OTUs identified in anaer-obic digesters. Systematic studies examining multiple AD reactordesigns with greater depth of coverage should help further definethe ‘core microbiome’ in digesters.
Many of the sequences analyzed in this study were classified aseither uncultured bacteria or archaea. Although phylogenetic anal-ysis of 16S rRNA gene sequences can provide some insight into thefunctional diversity of anaerobic digesters, function-oriented ap-proaches are needed. Metagenomic studies, as well as techniquessuch as MAR-FISH and SIP, have been used to a limited degree todiscover the function of uncultured bacterial groups (Ariesyadyet al., 2007a; Schlüter et al., 2008). While these methods shouldprobably be used more frequently, ultimately cultivation-basedstudies are also needed to define the functions of uncharacterizedgroups of bacteria and archaea in anaerobic digesters.
3.5. Analysis considerations
Analysis of a composite dataset carries a few potential caveatsand limitations that bear mentioning. While datasets from manydifferent studies were combined in this study, the single datasetby Rivière et al. (2009) was the source of over 50% of the sequencesanalyzed. Furthermore, the two large datasets created by Chouariet al. (2003, 2005) both generated sequences from one of the samesources used by Rivière, a municipal WWTP plant digester in Evry,France. These three studies have the effect of artificially inflatingthe representation of digesters handling municipal sludge com-pared to other types of AD systems. Additionally, some studies fo-cused solely on one or a few phylogenetic groups, again skewingthe composite dataset by increasing sequences from these groups.This case is most exemplified by the study by Chouari et al. (2003)where Planctomyces-specific PCR primers were used. As such, whilethe global diversity defined by the dataset can represent the typesof bacteria and archaea generally present in a digester, the abun-dance of some OTUs may not directly reflect the actual abundanceof the corresponding organisms found in any given AD system.
A second issue is related to the decision, made by researchers,as to which sequences (all vs. unique) to submit to public

3738 M.C. Nelson et al. / Bioresource Technology 102 (2011) 3730–3739
databases and how to describe these sequences. One third of thesequences in the composite dataset were unpublished at the timethey were accessed, and thus had little associated metadataaccompanying them. These two issues become important whendetermining maximum species richness or assessing the currentlevel of coverage and hinders determination of the beta diversityfound between different digesters. As previously suggested bySchloss and Handelsman (2004), standardized criteria for thesubmission of sequences to public databases could help improvethe quality of data available to researchers working in the AD field.As a beginning to this discussion, it is proposed that researchersinvestigating AD systems submit all high-quality sequences theyhave generated to the public databases (GenBank/EMBL/DDJ) afterthorough chimera checking. The resulting increase in sequenceredundancy will greatly increase the confidence of maximum rich-ness estimates. Information on key parameters, especially reactordesign, feedstock, and operational temperature, should also beincluded.
4. Conclusions
Nearly 90% of the archaeal and 60% of the bacterial species-leveldiversity in anaerobic digestion (AD) systems has been witnessed.Sequences from the bacterial phyla Chloroflexi, Proteobacteria,Firmicutes, Bacteroidetes and archaeal class Methanomicrobia arewell represented by the available sequences and the correspondingmicroorganisms are probably important participants in the ADprocess. The global diversity contains numerous groups for whichthere is no close cultured representative, especially the majorityof sequences assigned to the phyla Chloroflexi and Bacteroidetes. Fu-ture studies will need to utilize multiple approaches to furthercharacterize the microbial diversity and its function in individualAD systems.
Acknowledgement
This work was partially supported by a Department of Energygrant (DE-FG36-05GO85010).
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.biortech.2010.11.119.
References
Ariesyady, H.D., Ito, T., Okabe, S., 2007a. Functional bacterial and archaealcommunity structures of major trophic groups in a full-scale anaerobic sludgedigester. Water Res. 41, 1554–1568.
Ariesyady, H.D., Ito, T., Yoshiguchi, K., Okabe, S., 2007b. Phylogenetic and functionaldiversity of propionate-oxidizing bacteria in an anaerobic digester sludge. Appl.Microbiol. Biotechnol. 75, 673–683.
Blackall, L.L., Stratton, H., Bradford, D., Dot, T.D., Sjörup, C., Seviour, E.M., Seviour,R.J., 1996. ‘‘Candidatus Microthrix parvicella’’, a filamentous bacterium fromactivated sludge sewage treatment plants. Int. J. Syst. Bacteriol. 46, 344–346.
Burrell, P.C., O’Sullivan, C., Song, H., Clarke, W.P., Blackall, L.L., 2004. Identification,detection, and spatial resolution of Clostridium populations responsible forcellulose degradation in a methanogenic landfill leachate bioreactor. Appl.Environ. Microbiol. 70, 2414–2419.
Chen, S., Dong, X., 2005. Proteiniphilum acetatigenes gen. nov., sp. nov., from a UASBreactor treating brewery wastewater. Int. J. Syst. Evol. Microbiol. 55, 2257–2261.
Chen, Y., Cheng, J.J., Creamer, K.S., 2008. Inhibition of anaerobic digestion process: areview. Bioresour. Technol. 99, 4044–4064.
Chouari, R., Le Paslier, D., Daegelen, P., Ginestet, P., Weissenbach, J., Sghir, A., 2003.Molecular evidence for novel planctomycete diversity in a municipalwastewater treatment plant. Appl. Environ. Microbiol. 69, 7354–7363.
Chouari, R., Le Paslier, D., Daegelen, P., Ginestet, P., Weissenbach, J., Sghir, A., 2005.Novel predominant archaeal and bacterial groups revealed by molecularanalysis of an anaerobic sludge digester. Environ. Microbiol. 7, 1104–1115.
DeSantis, T.Z., Hugenholtz, P., Keller, K., Brodie, E.L., Larsen, N., Piceno, Y.M.,Phan, R., Andersen, G.L., 2006. NAST: a multiple sequence alignment serverfor comparative analysis of 16S rRNA genes. Nucleic Acids Res. 34, W394–W399.
Figuerola, E.L.M., Erijman, L., 2007. Bacterial taxa abundance pattern in an industrialwastewater treatment system determined by the full rRNA cycle approach.Environ. Microbiol. 9, 1780–1789.
Ganesan, A., Chaussonnerie, S., Tarrade, A., Dauga, C., Bouchez, T., Pelletier, E., LePaslier, D., Sghir, A., 2008. Cloacibacillus evryensis gen. nov., sp. nov., a novelasaccharolytic, mesophilic, amino-acid-degrading bacterium within the phylum‘Synergistetes’, isolated from an anaerobic sludge digester. Int. J. Syst. Evol.Microbiol. 58, 2003–2012.
Godon, J.J., Zumstein, E., Dabert, P., Habouzit, F., Moletta, R., 1997. Molecularmicrobial diversity of an anaerobic digestor as determined by small-subunitrDNA sequence analysis. Appl. Environ. Microbiol. 63, 2802–2813.
Hernon, F., Forbes, C., Colleran, E., 2006. Identification of mesophilic andthermophilic fermentative species in anaerobic granular sludge. Water Sci.Technol. 54, 19–24.
Holmes, D.E., Nevin, K.P., Woodard, T.L., Peacock, A.D., Lovley, D.R., 2007.Prolixibacter bellariivorans gen. nov., sp. nov., a sugar-fermenting,psychrotolerant anaerobe of the phylum Bacteroidetes, isolated from amarine-sediment fuel cell. Int. J. Syst. Evol. Microbiol. 57, 701–707.
Hori, T., Haruta, S., Ueno, Y., Ishii, M., Igarashi, Y., 2006. Dynamic transition of amethanogenic population in response to the concentration of volatile fatty acidsin a thermophilic anaerobic digester. Appl. Environ. Microbiol. 72, 1623–1630.
Hughes, J.B., Hellmann, J.J., Ricketts, T.H., Bohannan, B.J., 2001. Counting theuncountable: statistical approaches to estimating microbial diversity. Appl.Environ. Microbiol. 67, 4399–4406.
Krause, L., Diaz, N.N., Edwards, R.A., Gartemann, K.-H., Krömeke, H., Neuweger, H.,Pühler, A., Runte, K.J., Schlüter, A., Stoye, J., Szczepanowski, R., Tauch, A.,Goesmann, A., 2008. Taxonomic composition and gene content of a methane-producing microbial community isolated from a biogas reactor. J. Biotechnol.136, 91–101.
Kröber, M., Bekel, T., Diaz, N.N., Goesmann, A., Jaenicke, S., Krause, L., Miller, D.,Runte, K.J., Viehöver, P., Pühler, A., Schlüter, A., 2009. Phylogeneticcharacterization of a biogas plant microbial community integrating clonelibrary 16S-rDNA sequences and metagenome sequence data obtained by 454-pyrosequencing. J. Biotechnol. 142, 38–49.
Kurahashi, M., Fukunaga, Y., Sakiyama, Y., Harayama, S., Yokota, A., 2009. Iamiamajanohamensis gen. nov., sp. nov., an actinobacterium isolated from seacucumber Holothuria edulis, and proposal of Iamiaceae fam. nov.. Int. J. Syst.Evol. Microbiol. 59, 869–873.
Larue, R., Yu, Z., Parisi, V.A., Egan, A.R., Morrison, M., 2005. Novel microbial diversityadherent to plant biomass in the herbivore gastrointestinal tract, as revealed byribosomal intergenic spacer analysis and rrs gene sequencing. Environ.Microbiol. 7, 530–543.
Leclerc, M., Delgenès, J.-P., Godon, J.-J., 2004. Diversity of the archaeal community in44 anaerobic digesters as determined by single strand conformationpolymorphism analysis and 16S rDNA sequencing. Environ. Microbiol. 6, 809–819.
Lefebvre, O., Quentin, S., Torrijos, M., Godon, J.J., Delgenès, J.-P., Moletta, R., 2007.Impact of increasing NaCl concentrations on the performance and communitycomposition of two anaerobic reactors. Appl. Microbiol. Biotechnol. 75, 61–69.
Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., YadhukumarBuchner, A.,Lai, T., Steppi, S., Jobb, G., Förster, W., Brettske, I., Gerber, S., Ginhart, A.W., Gross,O., Grumann, S., Hermann, S., Jost, R., König, A., Liss, T., Lüssmann, R., May, M.,Nonhoff, B., Reichel, B., Strehlow, R., Stamatakis, A., Stuckmann, N., Vilbig, A.,Lenke, M., Ludwig, T., Bode, A., Schleifer, K.-H., 2004. ARB: a softwareenvironment for sequence data. Nucleic Acids Res. 32, 1363–1371.
Malin, C., Illmer, P., 2008. Ability of DNA content and DGGE analysis to reflect theperformance condition of an anaerobic biowaste fermenter. Microbiol. Res. 163,503–511.
Rivière, D., Desvignes, V., Pelletier, E., Chaussonnerie, S., Guermazi, S., Weissenbach,J., Li, T., Camacho, P., Sghir, A., 2009. Towards the definition of a core ofmicroorganisms involved in anaerobic digestion of sludge. ISME J. 3, 700–714.
Schloss, P.D., Handelsman, J., 2004. Status of the microbial census. Microbiol. Mol.Biol. Rev. 68, 686–691.
Schloss, P.D., Westcott, S.L., Ryabin, T., Hall, J.R., Hartmann, M., Hollister, E.B.,Lesniewski, R.A., Oakley, B.B., Parks, D.H., Robinson, C.J., Sahl, J.W., Stres, B.,Thallinger, G.G., Van Horn, D.J., Weber, C.F., 2009. Introducing mothur: open-source, platform-independent, community-supported software for describingand comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541.
Schlüter, A., Bekel, T., Diaz, N.N., Dondrup, M., Eichenlaub, R., Gartemann, K.-H.,Krahn, I., Krause, L., Krömeke, H., Kruse, O., Mussgnug, J.H., Neuweger, H.,Niehaus, K., Pühler, A., Runte, K.J., Szczepanowski, R., Tauch, A., Tilker, A.,Viehöver, P., Goesmann, A., 2008. The metagenome of a biogas-producingmicrobial community of a production-scale biogas plant fermenter analysed bythe 454-pyrosequencing technology. J. Biotechnol. 136, 77–90.
Sousa, D.Z., Pereira, M.A., Smidt, H., Stams, A.J.M., Alves, M.M., 2007. Molecularassessment of complex microbial communities degrading long chain fatty acidsin methanogenic bioreactors. FEMS Microbiol. Ecol. 60, 252–265.
Speece, R.E., Boonyakitsombut, S., Kim, M., Azbar, N., Ursillo, P., 2006. Overview ofanaerobic treatment: thermophilic and propionate implications. Water Environ.Res. 78, 460–473.

M.C. Nelson et al. / Bioresource Technology 102 (2011) 3730–3739 3739
Ueki, A., Akasaka, H., Suzuki, D., Ueki, K., 2006. Paludibacter propionicigenes gen. nov.,sp. nov., a novel strictly anaerobic, Gram-negative, propionate-producingbacterium isolated from plant residue in irrigated rice-field soil in Japan. Int.J. Syst. Evol. Microbiol. 56, 39–44.
Wang, Q., Garrity, G.M., Tiedje, J.M., Cole, J.R., 2007. Naive Bayesian classifier forrapid assignment of rRNA sequences into the new bacterial taxonomy. Appl.Environ. Microbiol. 73, 5261–5267.
Youssef, N.H., Elshahed, M.S., 2008. Species richness in soil bacterial communities: aproposed approach to overcome sample size bias. J. Microbiol. Methods 75, 86–91.
Yu, Z., Morrison, M., Schanbacher, F.L., 2010. Production and utilization of methanebiogas as renewable fuel. In: Alain Vertes, N.Q., Yukawa, Hideaki., Blaschek,Hans. (Eds.), Biomass to Biofuels: Strategies for Global Industries. Wiley,New York.
Zhang, H., Banaszak, J.E., Parameswaran, P., Alder, J., Krajmalnik-Brown, R.,Rittmann, B.E., 2009. Focused-Pulsed sludge pre-treatment increases thebacterial diversity and relative abundance of acetoclastic methanogens in afull-scale anaerobic digester. Water Res. 43, 4517–4526.