a few basics about qtl mapping
TRANSCRIPT

1
A few FAQs About QTL
Mapping
Guoyou Ye
E-mail: [email protected]
Basics of QTL Mapping
What is QTL Mapping?
Map individual genetic factors on the quantitative traits,
to specific chromosomal segments in the genome.
The key questions in QTL mapping studies are:
� How many QTL are there?
� Where are they in the marker map?
� How large an influence does each of them have on
the trait of interest?
Dataset of QTL Mapping
� Mapping population
� Linkage map (can be constructed
using your own marker genotype
data)
� Marker genotype
� Phenotypic data

Mapping Population
� Biparental populations
� F2
� BC: backcross
� DH, doubled haploids
� RIL, recombination inbred lines
� CSSL: chromosome segment substitution
lines
� Natural population
Maximum Likelihood Method for
Estimating Recombination Rate
Recombination Rate
� Recombinant gametes: are derived by crossing over between the two parental gametes received by an individual.
� Recombination fraction: is the probability that gamete transmitted by an individual is recombinant.
Expected Marker Genotype Frequencies
BC1 BC2 DH Counts Expected Freq
M1M1M2M2 M1m1M2m2 M1M1M2M2 n1 f1= 21 (1----r)
M1M1M2m2 M1m1m2m2 m1m1M2M2 n2 f2= 21 r
M1m1M2M2 m1m1M2m2 m1m1M2M2 n3 f3= 21 r
M1m1M2m2 m1m1m2m2 m1m1m2m2 n4 f4= 21 (1----r)

Estimating Recombination Rate
� Likelihood function
� Logarithm operation of the likelihood function
� Maximum likelihood estimate
� Information: The negatives of the second derivatives
4321 )]1([][][)]1([!!!!
!21
21
21
21
4321
nnnnrrrr
nnnn
nL −−=
rnnrnnCL ln)()1ln()(lnln 3241 ++−++=
n
nn
nnnn
nnr 32
4321
32ˆ+
=+++
+=
)1(]
)1([)
ln(
2
32
2
41
2
2
rr
n
r
nn
r
nnE
rd
LdEI
−=
+−
−
+−−=−=
n
rr
IVr
)ˆ1(ˆ1ˆ
−==
Example
� BC experiment
� P1 and P2 are AABB and aabb, respectively
� Number of plants of different genotypes in the population
� AABB:162;AABb:40;AaBB:41;AaBb:158
%20.20401
81
1584140162
4140ˆ ==
+++
+=r
4
ˆ 1002.4)ˆ1(ˆ −
×≈−
=n
rrVr
Recombination Rate When Three
Markers Are Linked
�Whenδ=0 (Crossovers in the two intervals are
independent),
or
�when δ= 1 (Complete interference, crossover at on
interval completely prevent crossover event in the other
interval)
2312231213 )1(2 rrrrr δ−−+=
2312231213 )1)(1()1( rrrrr +−−=−
231223122312231213 2)1()1( rrrrrrrrr −+=−+−=
231213 rrr +=
Mapping Function
� Mapping distance
� Unit of mapping distance:Morgan (M)or centi-Morgan (cM),
1M= 100 cM
� Morgan is defined as that distance along which one crossing over is
expected to occur per gamete per generation
� Mapping distance m is function of recombination rate:
� f is called mapping function
231213 mmm +=
)(rfm =

Common Mapping Functions
• Morgan
m =r ×100 (cM)
• Haldane: no interference
• Kosambi with interference
)1( 2
21 mer −
−=
r
rm
21
21ln25
−
+=
1
1
2
125/
25/
+
−=
ee
m
m
r
三种作图函数的比较
15
Basic Principle of QTLMapping
� Distribution of trait values of three marker genotypes of a maker
Maximum likelihood Estimation
� A common approach in statistical estimation
• Define hypotheses
• Generate likelihood function
• Estimate
• Test hypotheses
• Draw statistical conclusions

Hypotheses in Linkage Analysis
� H0:
� θ = 0.5
� the QTL is not linked to the marker(s)
� HA:
� θ ≠ 0.5
� the QTL is linked to the marker(s)
Expected Marker Genotype
Frequencies
BC1 BC2 DH Counts Expected Freq
M1M1M2M2 M1m1M2m2 M1M1M2M2 n1 f1= 21 (1----r)
M1M1M2m2 M1m1m2m2 m1m1M2M2 n2 f2= 21 r
M1m1M2M2 m1m1M2m2 m1m1M2M2 n3 f3= 21 r
M1m1M2m2 m1m1m2m2 m1m1m2m2 n4 f4= 21 (1----r)
QTL Effects and QTL Genotypic
Value
� The additive (a) and dominance (d) genetic model
� P1 (A1A1): m+a; F1 (A1A2): m+d; P2 (A2A2): m-a
� Code of the three genotypes at a locus: 2 (P1), 0 (P2), 1 (F1)
Genotype P2: A2A2 (0) F1: A1A2 (1) P1: A1A1 (2)
mid-parent m
Genotypic G22 G12 G11
value
Additive a Additive a
Dominance d20
Quiz 1::::What is LOD ????
� LRT: likelihood ratio test
� LOD : (Likelihood of Odd)
� A LOD-score threshold of 3 corresponds to a single-test p-value of
approximately 0.0001
)ln(2 0
AL
LLRT −=
)log()log()log( 0
0
LLL
LLOD A
A−==
61.4)10ln(2
LRTLRTLOD ≈= LODLRT 61.4≈
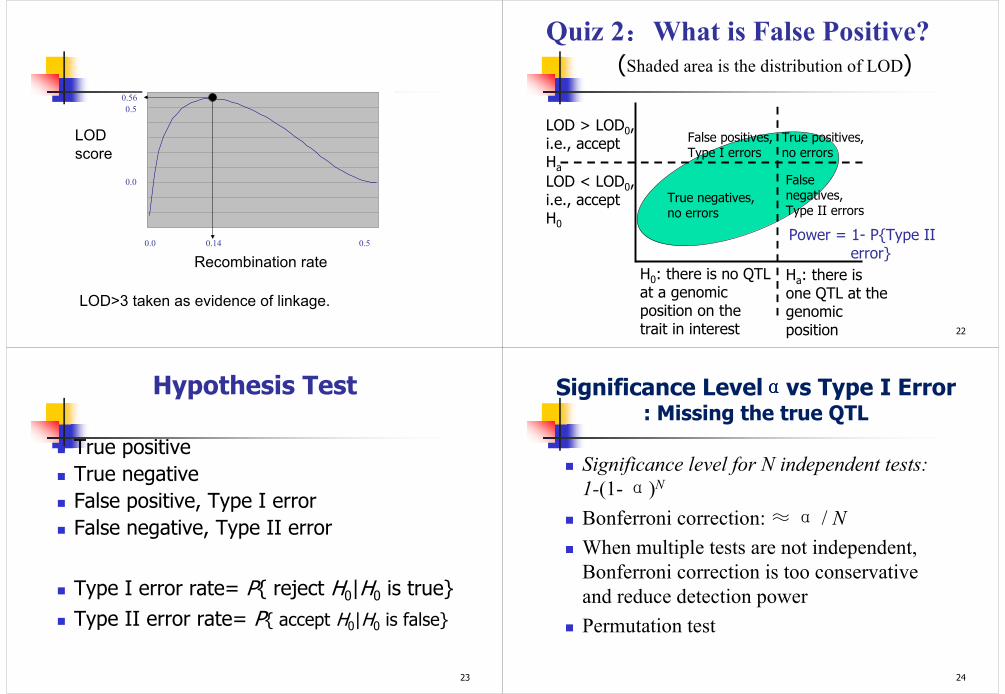
0.0 0.5
0.0
0.5
0.14
0.56
Recombination rate
LOD
score
LOD>3 taken as evidence of linkage.
22
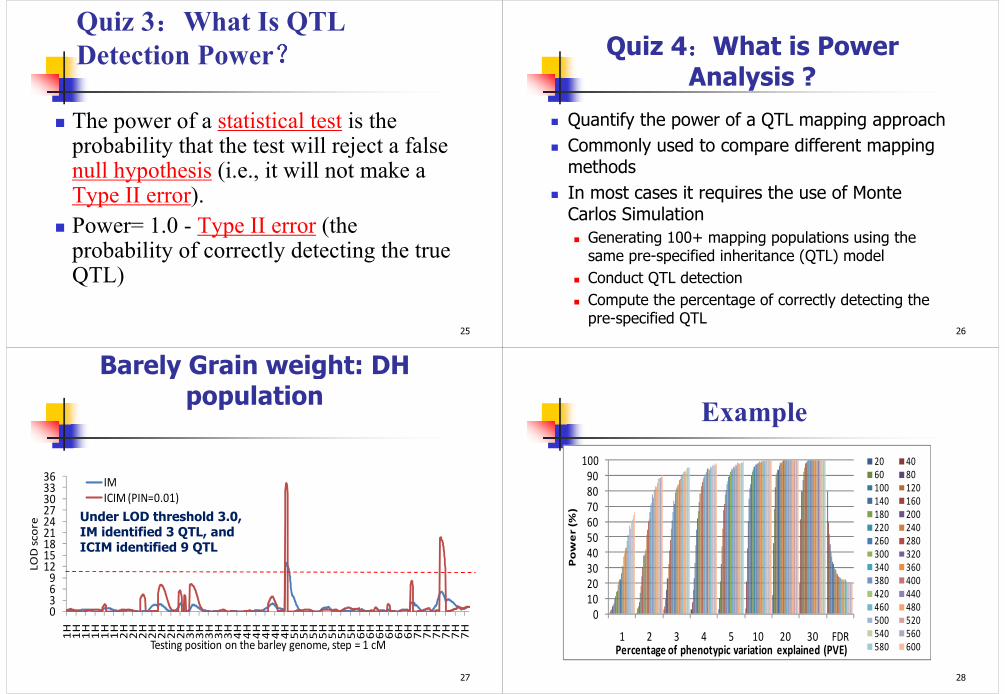
Quiz 2::::What is False Positive?(Shaded area is the distribution of LOD)
H0: there is no QTL at a genomic position on the trait in interest
Ha: there is one QTL at the genomic position
LOD < LOD0, i.e., accept H0
LOD > LOD0, i.e., accept Ha
True negatives, no errors
True positives, no errors
False positives, Type I errors
False negatives, Type II errors
Power = 1- P{Type II error}
23
Hypothesis Test
� True positive
� True negative
� False positive, Type I error
� False negative, Type II error
� Type I error rate= P{ reject H0|H0 is true}
� Type II error rate= P{ accept H0|H0 is false}
Significance Levelααααvs Type I Error : Missing the true QTL
� Significance level for N independent tests:
1-(1- α)N
� Bonferroni correction: ≈α / N
� When multiple tests are not independent,
Bonferroni correction is too conservative
and reduce detection power
� Permutation test
24
25
Quiz 3::::What Is QTL
Detection Power????
� The power of a statistical test is the probability that the test will reject a false null hypothesis (i.e., it will not make a Type II error).
� Power= 1.0 - Type II error (the probability of correctly detecting the true QTL)
Quiz 4::::What is Power Analysis ?
� Quantify the power of a QTL mapping approach
� Commonly used to compare different mapping methods
� In most cases it requires the use of Monte Carlos Simulation
� Generating 100+ mapping populations using the same pre-specified inheritance (QTL) model
� Conduct QTL detection
� Compute the percentage of correctly detecting the pre-specified QTL
26
27
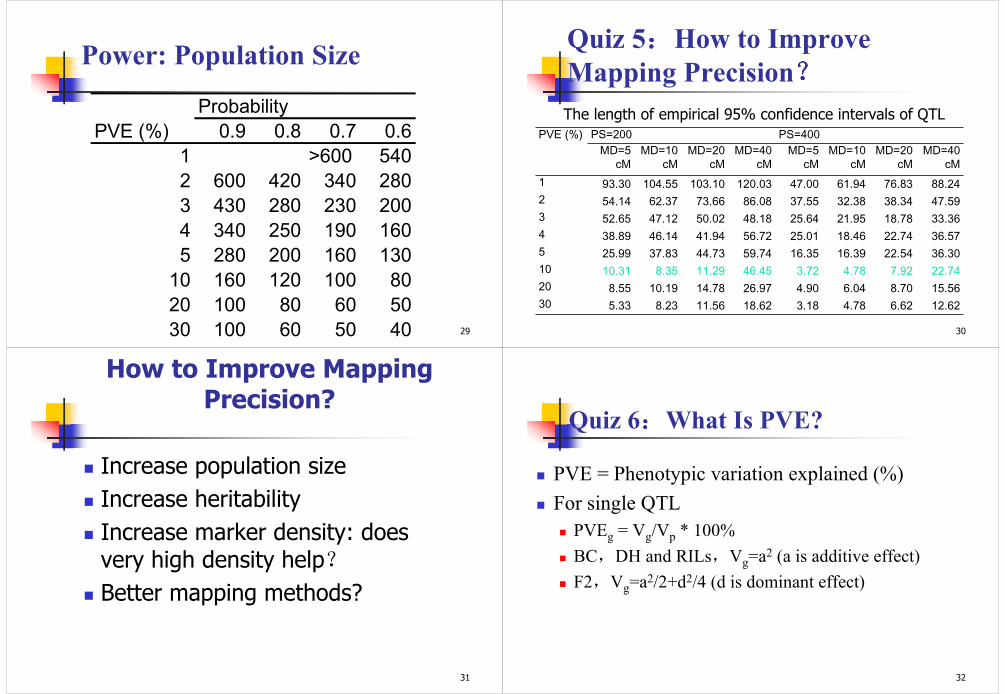
Barely Grain weight: DH population
0369
121518212427303336
1H
1H
1H
1H
1H
1H
2H
2H
2H
2H
2H
2H
2H
3H
3H
3H
3H
3H
4H
4H
4H
4H
4H
4H
5H
5H
5H
5H
5H
5H
5H
6H
6H
6H
6H
6H
6H
7H
7H
7H
7H
7H
7H
LO
D s
co
re
Testing position on the barley genome, step = 1 cM
IM
ICIM (PIN=0.01)
Under LOD threshold 3.0, IM identified 3 QTL, and ICIM identified 9 QTL
Example
28
0
10
20
30
40
50
60
70
80
90
100
1 2 3 4 5 10 20 30 FDR
Po
we
r (
%)
Percentage of phenotypic variation explained (PVE)
20 40
60 80
100 120
140 160
180 200
220 240
260 280
300 320
340 360
380 400
420 440
460 480
500 520
540 560
580 600
Power: Population Size
29
Probability
PVE (%) 0.9 0.8 0.7 0.6
1 >600 540
2 600 420 340 280
3 430 280 230 200
4 340 250 190 160
5 280 200 160 130
10 160 120 100 80
20 100 80 60 50
30 100 60 50 40
Quiz 5::::How to Improve
Mapping Precision????
30
PVE (%) PS=200 PS=400MD=5
cMMD=10
cMMD=20
cMMD=40
cMMD=5
cMMD=10
cMMD=20
cMMD=40
cM1 93.30 104.55 103.10 120.03 47.00 61.94 76.83 88.242 54.14 62.37 73.66 86.08 37.55 32.38 38.34 47.593 52.65 47.12 50.02 48.18 25.64 21.95 18.78 33.364 38.89 46.14 41.94 56.72 25.01 18.46 22.74 36.575 25.99 37.83 44.73 59.74 16.35 16.39 22.54 36.3010 10.31 8.35 11.29 46.45 3.72 4.78 7.92 22.7420 8.55 10.19 14.78 26.97 4.90 6.04 8.70 15.5630 5.33 8.23 11.56 18.62 3.18 4.78 6.62 12.62
The length of empirical 95% confidence intervals of QTL
How to Improve Mapping Precision?
� Increase population size
� Increase heritability
� Increase marker density: does very high density help?
� Better mapping methods?
31 32
Quiz 6::::What Is PVE?
� PVE = Phenotypic variation explained (%)
� For single QTL
� PVEg = Vg/Vp * 100%
� BC,DH and RILs,Vg=a2 (a is additive effect)
� F2,Vg=a2/2+d2/4 (d is dominant effect)

Quiz 7::::Can We Add PVEs for
Each of the QTL Together?
� Z=X+Y, X and Y are random variables
� E(X+Y) = E(X) + E(Y)
� V(X+Y) = V(X) + V(Y) +2Cov(X, Y)
� When all QTL are independent, Cov(X, Y)=0,
PVEs can be added together to measure
totally how much varinace is accounted for.
� When QTL are linked, overall PVE does not
equal to the sum of individual PVEs
33
Quiz 8::::Can the Overall PVE Be
More Than 100%?
� In RIL,locus A, genetic variance is a12 while for locus B
it is a22. the total genetic variance is a1
2 +a22+2(1-2r)a1a2
� Thus, when QTL are linked, total PVE can be higher than
100%; But, for unlinked QTL PVE cannot be higher than
100%
34
基因型 频率 基因型值
AABB )1(2
1 r− a1+a2
AAbb r2
1 a1-a2
aaBB r2
1 -a1+a2
aabb )1(2
1 r− -a1-a2
Quiz 9::::What is Distorted
Segregation?
� Theoretical segregation ratio
� P1 (AA) X P2 (aa) (no selection)
� P1BC1: AA:Aa=1:1
� P2BC1: Aa:aa=1:1
� F2: AA:Aa:aa=1:2:1
� DH, RIL: AA:aa=1:1
� Segregation Distortion
� Random drift
� Gamete/ zygote selection
35
Segregation Distortion in An
Actual Rice F2 Population
RP178
RM143
RM159
RM44 RM304
RM147
RM552
RP129
RM491
0
4
8
12
16
20
1 1 1 1 2 2 2 2 3 3 3 3 4 4 5 5 5 5 6 6 6 6 7 7 8 8 9 9
10
10
11
11
11
12
12
significance level (-log10P)
Markers on the 12 rice chromosomes

When Distorted Markers Are Not
Linked With QTLA
B
0
20
40
60
80
100
qPH1-1 qPH1-2 qPH3-1 qPH3-2 qPH4 qPH5 qPH6 qPH7 qPH12 FDR
Power (%)
No distortion SDM1 SDM2 SDM3SDM4 SDM5 SDM6 SDM7SDM8 SDM9
0
20
40
60
80
100
qHD1 qHD3 qHD4 qHD5 qHD8 qHD11 FDR
Power (%)
A,QTL for plant height, population size=180
B,QTL for heading days, population size=180
A
B
0
20
40
60
80
100
qPH1-1 qPH1-2 qPH3-1 qPH3-2 qPH4 qPH5 qPH6 qPH7 qPH12
Power (%)
No distortion SDM1 SDM2 SDM3
SDM4 SDM5 SDM6 SDM7
SDM8 SDM9
0
20
40
60
80
100
qPH1-1 qPH1-2 qPH3-1 qPH3-2 qPH4 qPH5 qPH6 qPH7 qPH12
Power (%)
C
D
0
20
40
60
80
100
qHD1 qHD3 qHD4 qHD5 qHD8 qHD11
Power (%)
0
20
40
60
80
100
120
qHD1 qHD3 qHD4 qHD5 qHD8 qHD11
Power (%)
When Distorted Markers Are
Linked With QTL
A,QTL for plant height, population size=180B,QTL for plant height, population size=500
C, QTL for heading days, population size=180D, QTL for heading days, population size=500
Effect of Segregation Distortion on
QTL Mapping
� If the distorted marker is not closely linked
with any plant height or heading date QTL, no
significant effects were observed on the
detection power.
� Otherwise, distorted marker may increase or
decrease the QTL detection power.
� In large-size populations, say size of 500, the
effect of distorted marker was minor even the
distorted marker was closely linked with QTL.
Quiz 10::::How to Handle
Missing Values ?
40
� Missing marker data
� Can be imputed using linkage
relationship
� Missing phenotypic data
� Replaced by population mean
� Delete the genotype
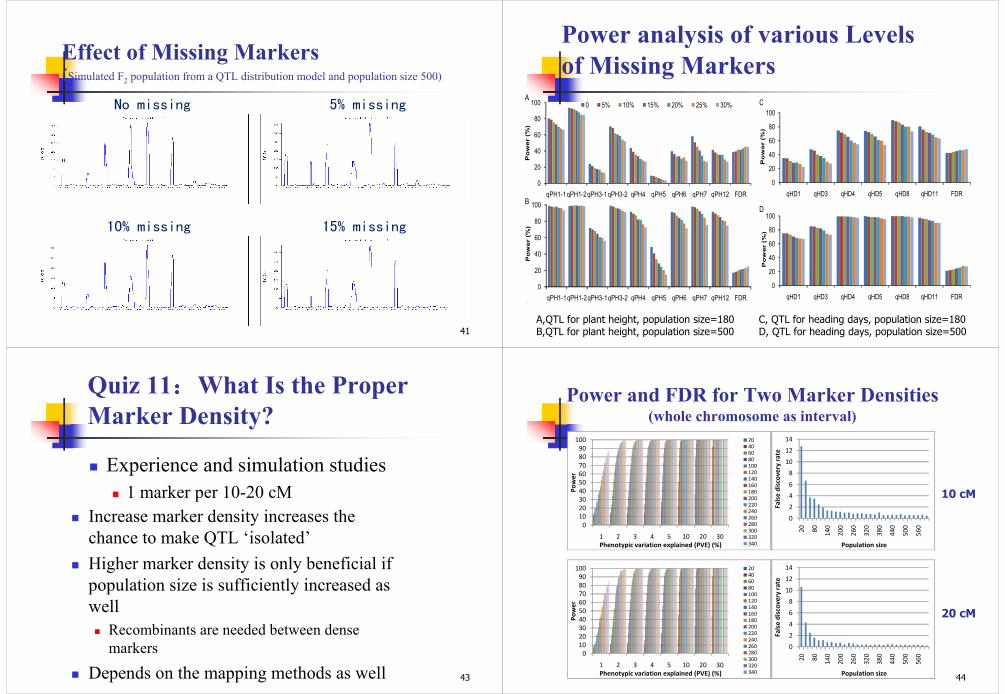
Effect of Missing Markers (Simulated F2 population from a QTL distribution model and population size 500)
41
No missing 5% missing
10% missing 15% missing
Power analysis of various Levels
of Missing MarkersA
B
C
0
20
40
60
80
100
qPH1-1qPH1-2qPH3-1qPH3-2 qPH4 qPH5 qPH6 qPH7 qPH12 FDR
Power (%)
0 5% 10% 15% 20% 25% 30%
0
20
40
60
80
100
qPH1-1qPH1-2qPH3-1qPH3-2 qPH4 qPH5 qPH6 qPH7 qPH12 FDR
Power (%)
C
D
0
20
40
60
80
100
qHD1 qHD3 qHD4 qHD5 qHD8 qHD11 FDR
Power (%)
0
20
40
60
80
100
qHD1 qHD3 qHD4 qHD5 qHD8 qHD11 FDR
Power (%)
A,QTL for plant height, population size=180B,QTL for plant height, population size=500
C, QTL for heading days, population size=180D, QTL for heading days, population size=500
Quiz 11::::What Is the Proper
Marker Density?
� Experience and simulation studies
� 1 marker per 10-20 cM
43
� Increase marker density increases the
chance to make QTL ‘isolated’
� Higher marker density is only beneficial if
population size is sufficiently increased as
well
� Recombinants are needed between dense
markers
� Depends on the mapping methods as well
Power and FDR for Two Marker Densities (whole chromosome as interval)
44
0
10
20
30
40
50
60
70
80
90
100
1 2 3 4 5 10 20 30
Po
we
r
Phenotypic variation explained (PVE) (%)
20
40
60
80
100
120
140
160
180
200
220
240
260
280
300
320
340
0
2
4
6
8
10
12
14
20
80
14
0
20
0
26
0
32
0
38
0
44
0
50
0
56
0
Fa
lse
dis
cov
ery
ra
te
Population size
0
10
20
30
40
50
60
70
80
90
100
1 2 3 4 5 10 20 30
Po
we
r
Phenotypic variation explained (PVE) (%)
20
40
60
80
100
120
140
160
180
200
220
240
260
280
300
320
340
0
2
4
6
8
10
12
14
20
80
14
0
20
0
26
0
32
0
38
0
44
0
50
0
56
0
Fa
lse
dis
cov
ery
ra
te
Population size
10 cM
20 cM

45
0
10
20
30
40
50
60
70
80
90
100
1 2 3 4 5 10 20 30
Po
we
r
Phenotypic variation explained (PVE) (%)
20
40
60
80
100
120
140
160
180
200
220
240
260
280
300
320
340
0
10
20
30
40
50
60
70
80
90
20
80
14
0
20
0
26
0
32
0
38
0
44
0
50
0
56
0
Fa
lse
dis
cov
ery
ra
te
Population size
0
10
20
30
40
50
60
70
80
90
100
1 2 3 4 5 10 20 30
Po
we
r
Phenotypic variation explained (PVE) (%)
20
40
60
80
100
120
140
160
180
200
220
240
260
280
300
320
340
0
10
20
30
40
50
60
70
80
90
20
80
14
0
20
0
26
0
32
0
38
0
44
0
50
0
56
0
Fa
lse
dis
cov
ery
ra
te
Population size
Power and FDR for Two Marker Densities(10cM interval, QTL in the middle)
10 cM
20 cM
Quiz 12::::What is QTL by E?
46
环境1 环境2
基因
型值
A. 互作模式1
基因型1
基因型2
环境1 环境2
基因型值
B. 互作模式2
基因型1
基因型2
环境1 环境2
基因型
值
C. 互作模式3
基因型1
基因型2
环境1 环境2
基因型
值
D. 互作模式4
基因型1
基因型2
47
Chromosome 1 2 3 3 5 5 7 7 7 8 8 8 9 10 12 12
Segment M4 M18 M21 M23 M35 M39 M49 M50 M51 M54 M56 M57 M59 M69 M77 M79
LODa E1 0.00 0.26 0.01 0.00 1.87 0.00 0.12 0.03 0.03 0.01 0.02 6.06 0.47 0.01 0.17 0.61
E2 0.00 1.05 2.88 4.66 0.95 0.14 0.35 0.30 0.11 0.13 5.84 3.92 2.89 0.22 0.12 0.05
E3 2.06 4.03 0.00 0.20 0.84 2.60 0.05 9.61 4.64 0.04 0.04 3.98 3.36 4.35 0.89 2.17
E4 0.04 0.04 0.00 4.03 0.05 0.32 0.04 0.02 0.49 0.70 0.05 15.89 6.26 2.57 0.10 0.24
E5 0.35 0.66 0.41 3.13 0.40 0.00 4.09 12.51 16.53 2.04 3.18 5.56 9.04 0.31 0.42 0.15
E6 0.13 0.38 1.06 1.35 0.30 0.22 0.32 0.02 0.18 0.00 0.16 1.84 4.15 0.90 0.29 0.05
E7 0.08 5.07 0.22 0.21 0.04 0.26 1.47 0.33 0.07 0.03 5.52 6.33 4.07 1.66 4.04 0.01
E8 0.01 0.00 0.02 0.45 2.16 1.54 0.01 0.02 0.01 0.67 0.17 16.89 10.01 0.01 0.00 0.00
ADDb E1 -0.11 -0.72 -0.12 -0.08 -2.13 0.06 -0.36 -0.19 0.25 -0.13 0.17 4.13 1.27 0.08 -0.35 -1.99
E2 -0.07 2.21 -2.98 4.35 -2.11 0.78 -0.88 -0.89 -0.72 0.54 4.43 4.51 4.62 -0.72 -0.43 -0.78
E3 3.89 4.69 0.11 -0.84 -2.03 -3.66 0.32 -6.12 5.11 -0.32 0.35 4.62 5.16 3.57 -1.21 5.68
E4 -0.24 -0.23 -0.04 2.05 -0.25 -0.61 -0.16 0.12 0.76 -0.67 -0.18 5.84 3.71 1.32 -0.20 -0.91
E5 1.06 1.22 -0.75 2.42 -0.94 0.07 2.30 -5.19 8.28 -1.59 2.20 3.86 6.40 0.60 -0.55 0.98
E6 -1.22 1.73 -2.29 2.88 -1.53 -1.30 -1.11 -0.30 -1.14 0.13 0.86 3.79 7.38 -1.87 -0.85 1.04
E7 -0.52 3.74 -0.56 0.61 -0.31 -0.78 -1.37 -0.69 -0.41 -0.19 3.11 4.29 4.01 1.44 -1.91 -0.21
E8 -0.09 0.05 -0.11 0.63 -1.63 -1.31 -0.09 -0.12 0.09 -0.62 -0.33 5.94 4.95 -0.09 0.03 0.09
PVEc E1 0.01 0.48 0.02 0.01 4.15 0.00 0.23 0.06 0.06 0.03 0.04 15.60 1.00 0.01 0.34 1.24
E2 0.00 2.24 6.60 11.44 2.04 0.28 0.69 0.59 0.24 0.26 14.54 9.36 6.66 0.46 0.25 0.10
E3 3.61 7.76 0.01 0.33 1.45 4.73 0.07 21.28 9.19 0.07 0.07 7.54 6.35 8.53 1.53 3.91
E4 0.04 0.05 0.00 5.62 0.06 0.38 0.05 0.03 0.59 0.86 0.05 34.66 9.49 3.35 0.11 0.29
E5 0.35 0.68 0.41 3.51 0.41 0.00 4.61 19.86 31.26 2.18 3.57 6.79 12.65 0.31 0.41 0.15
E6 0.46 1.37 3.88 4.99 1.07 0.78 1.07 0.07 0.60 0.02 0.55 6.60 16.93 3.06 0.98 0.17
E7 0.11 8.28 0.29 0.29 0.06 0.36 2.13 0.46 0.10 0.04 9.25 10.89 6.44 2.33 6.37 0.01
E8 0.01 0.00 0.02 0.52 2.66 1.70 0.02 0.02 0.01 0.73 0.17 35.08 16.53 0.01 0.00 0.00
Chromosome segments showing
QTL for ACEQuiz 13::::Does the Phenotypic Data Need to
Follow Normal Distribution?
48
F2h mg
2=0.90 , h p g
2=0 .10
B 1h mg
2=0.00
h pg2=1.00
B2h mg
2=0.95 , h p g
2=0 .05
F2 :3h m g
2=0.89 , h pg
2=0.11
R ILh m g
2=0 .88 , h pg
2=0 .12
F2h mg
2=0.80 , h p g
2=0 .10
B1h m g
2=0 .00
h p g2=0 .38
B2h mg
2=0 .87 , h p g
2=0 .05
F2 :3h m g
2=0 .82
h p g2=0 .12
R ILh m g
2=0 .84 , h pg
2=0 .13
F2h mg
2=0.70 , h p g
2=0 .10
B1h m g
2=0.00
h pg2=0 .23
B2h mg
2=0.78 , h pg
2=0.05
F2 :3h m g
2=0 .74
h p g2=0 .12
R ILh m g
2=0 .78 , h pg
2=0 .13
F2h mg
2=0.60 , h p g
2=0 .10
B1h m g
2=0 .00
h p g2=0 .17
B2h m g
2=0 .69 , h pg
2=0 .05
F2 :3h m g
2=0 .66
h p g2=0 .12
R ILh m g
2=0 .72 , h pg
2=0 .14
F2h mg
2=0.50 , h p g
2=0 .10
B1h m g
2=0 .00
h p g2=0 .13
B2h m g
2=0 .59 , h pg
2=0 .05
F2 :3h m g
2=0 .57
h p g2=0 .13
R ILh m g
2=0 .65 , h pg
2=0 .16
F2h mg
2=0.40 , h p g
2=0 .10
B1h mg
2=0.00
h pg2=0.11
B2h m g
2=0 .49 , h pg
2=0 .05
F2 :3h m g
2=0 .47
h p g2=0 .13
R ILh m g
2=0 .57 , h pg
2=0 .17
F2h mg
2=0.30 , h p g
2=0 .10
B1h mg
2=0.00
h pg2=0.09
B2h m g
2=0.38
h pg2=0 .06
F2 :3h m g
2=0 .37
h p g2=0 .14
R ILh m g
2=0 .47 , h pg
2=0 .19
F2h m g
2=0 .20
h p g2=0 .10
B1h mg
2=0.00
h pg2=0.08
B2h m g
2=0.26
h pg2=0 .06
F2 :3h m g
2=0 .26
h p g2=0 .15
R ILh m g
2=0 .34
h pg2=0.21
A single major gene and
many minor genes: the
distribution will be
normal.
However, the residual
needs to be normally
distributed.
There are methods that
can handle non-normal
distribution of residual.

A simulation: A QTL accounted for 80% of the phenotypic variation with additive effect 1.0
and located at 25 cM
49
0
10
20
30
40
50
8.5 9 9.5 10 10.5 11 11.5 12
Fre
qu
en
cy
Phenotypic value
-0.2
0
0.2
0.4
0.6
0.8
1
1.2
0 51
01
52
02
53
03
54
04
55
05
56
06
57
07
58
08
59
09
51
00
10
51
10
11
51
20
12
51
30
13
51
40
14
51
50
15
51
60
Est
ima
ted
eff
ect
1D-scanning on one chromosome, step=1cM
ICIM
IM
0
20
40
60
80
100
0 51
01
52
02
53
03
54
04
55
05
56
06
57
07
58
08
59
09
51
00
10
51
10
11
51
20
12
51
30
13
51
40
14
51
50
15
51
60
LO
D s
co
re
1D-scanning on one chromosome, step=1cM
ICIM
IM
Quiz 14::::Do Mapping Component Traits Help?
50
Effect Trait I Trait II Addition Subtraction Multiplication Division
Mean 25 20 45 5 500 1.2563
A1 1 0 1 1 20 0.0503
A2 1 0 1 1 20 0.0503
A3 0 1 1 -1 25 -0.0631
A4 0 1 1 -1 25 -0.0631
A12 0 0 0 0 0 0
A13 0 0 0 0 1 -0.0025
A14 0 0 0 0 1 -0.0025
A23 0 0 0 0 1 -0.0025
A24 0 0 0 0 1 -0.0025
A34 0 0 0 0 0 0.0063
A123 0 0 0 0 0 0
A124 0 0 0 0 0 0
A134 0 0 0 0 0 0.0003
A234 0 0 0 0 0 0.0003
A1234 0 0 0 0 0 0
Using Composite Trait Reduces
Detection Power and Increase FDR
51
QTL Trait I Trait II Addition Subtraction Multiplication Division
Model I Power (%) Q1 95.10 69.60 69.30 55.20 50.50
Q2 94.80 69.80 70.40 54.10 50.90
Q3 92.50 67.20 65.30 76.90 75.20
Q4 94.50 68.40 65.40 77.80 75.20
FDR (%) 21.63 22.98 27.42 28.05 28.07 29.68
Model II Power (%) Q1 95.40 67.40 65.60 54.80 49.90
Q2 92.90 62.40 66.00 50.00 49.90
Q3 93.70 69.90 67.00 79.20 74.90
Q4 91.90 62.40 64.90 73.50 72.90
FDR (%) 21.35 22.18 28.76 28.59 28.07 28.89
Model III Power (%) Q1 95.20 66.60 52.40 53.60 37.70
Q2 95.00 69.20 51.60 54.70 36.40
Q3 92.90 63.40 47.80 69.70 56.20
Q4 92.60 61.50 49.90 72.60 58.00
FDR (%) 19.78 23.44 28.83 27.71 29.74 30.18
Quiz 15: Is Selective Genotyping
(SG) Useful?
52L
LL
H
HH
LH
N
pp
N
pp
ppt
2
)1(
2
)1( −+
−
−=
Low tail
High tail

Comparison of SG with IM and ICIM(PVE= 5%, MD= 5cM and both tails have a selected proportion
of 10%)
SGM has higher detection power than the conventional IM but lower detection power
than ICIMSG may still be useful!