2. biosynthesis of natural products - terpene biosynthesis ... · biosynthesis of natural products...
TRANSCRIPT

1
2. Biosynthesis of Natural Products - Terpene Biosynthesis 2.1 Introduction Terpenes are a large and varied class of natural products, produced primarily by a wide variety of plants, insects, microoroganisms and animals. They are the major components of resin, and of turpentine produced from resin. The name "terpene" is derived from the word "turpentine". Terpenes are major biosynthetic building blocks within nearly every living creature. Steroids, for example, are derivatives of the triterpene squalene. When terpenes are modified, such as by oxidation or rearrangement of the carbon skeleton, the resulting compounds are generally referred to as terpenoids. Some authors will use the term terpene to include all terpenoids. Terpenoids are also known as Isoprenoids. Terpenes and terpenoids are the primary constituents of the essential oils of many types of plants and flowers. Essential oils are used widely as natural flavor additives for food, as fragrances in perfumery, and in traditional and alternative medicines such as aromatherapy. Synthetic variations and derivatives of natural terpenes and terpenoids also greatly expand the variety of aromas used in perfumery and flavors used in food additives. Recent estimates suggest that over 30'000 different terpenes have been characterized from natural sources. Early on it was recognized that the majority of terpenoid natural products contain a multiple of 5C-atoms. Hemiterpenes consist of a single isoprene unit, whereas the monoterpenes include e.g.:
CH2OH
CH2OH
OH
CHO
CHO O
O
Camphor!-Pinene
Citronellal
MentholCitralGeraniolNerolLimonensMyrcens
Monoterpenes
Terpenes with 15 C-atoms are known as sesquiterpenes :
CH2OH
O
Farnesol Bisabolene Cadinene Selinene Vetivone
HO
Patchoulol(Perfume)
O
COOH
OH Abscisic acid(Phytohormone)
O
O
O
COOMe
OH
Pentalenolactone(Antibiotic)
Sesquiterpenes
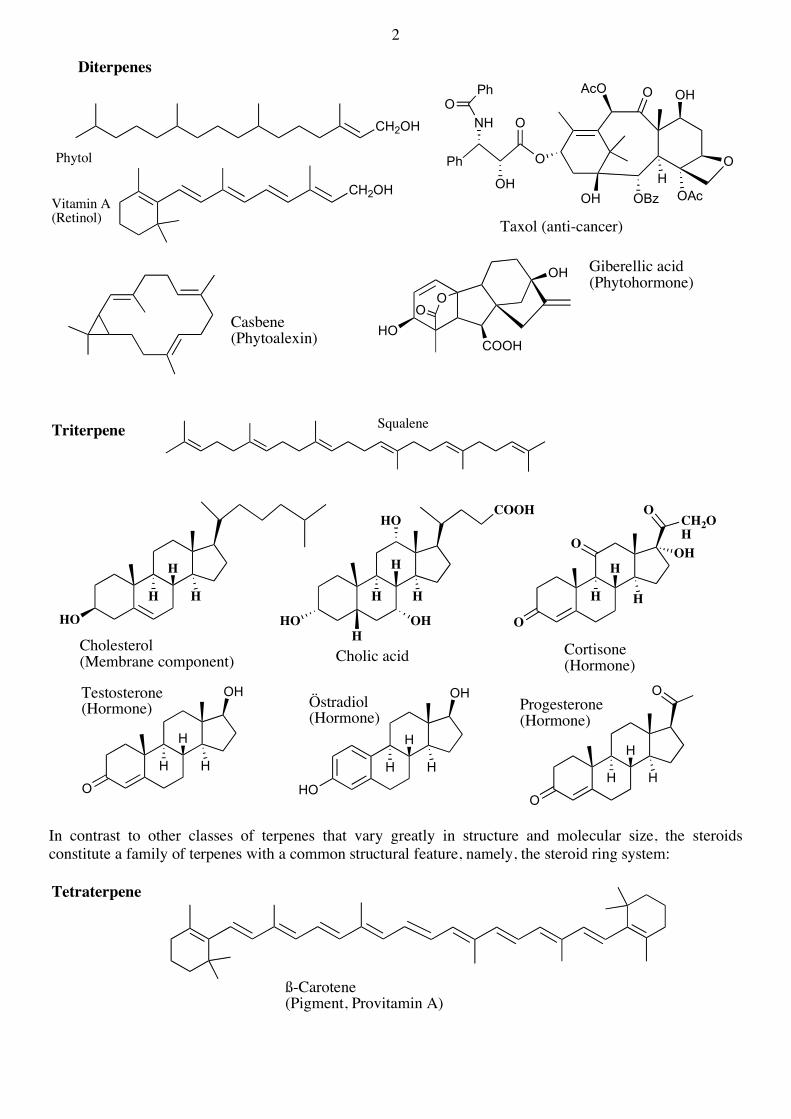
The terpenes containing, or originating from precursors, containing 20 C-atoms are known as diterpenes, those with 30 C-atoms as triterpenes and those with 40 C-atoms as tetraterpenes :
2
Diterpenes
CH2OH
CH2OH
Vitamin A(Retinol)
Phytol
AcO O OH
O
OAcOBzOH
OPh
O
OH
NH
O
Ph
H
Taxol (anti-cancer)
Casbene(Phytoalexin)
HO
O
COOH
Giberellic acid(Phytohormone)
OH
O
Triterpene
Squalene
HO
H
HH
HO
H
OH
H
HO
H
H
COOH
O
O
H
OH
HH
OCH2OH
Cholesterol(Membrane component) Cholic acid Cortisone
(Hormone)
H
O
OH
HH
H
HO
OH
HH
H
O
HH
OTestosterone(Hormone) Östradiol
(Hormone)Progesterone(Hormone)
In contrast to other classes of terpenes that vary greatly in structure and molecular size, the steroids constitute a family of terpenes with a common structural feature, namely, the steroid ring system: Tetraterpene
ß-Carotene(Pigment, Provitamin A)
3
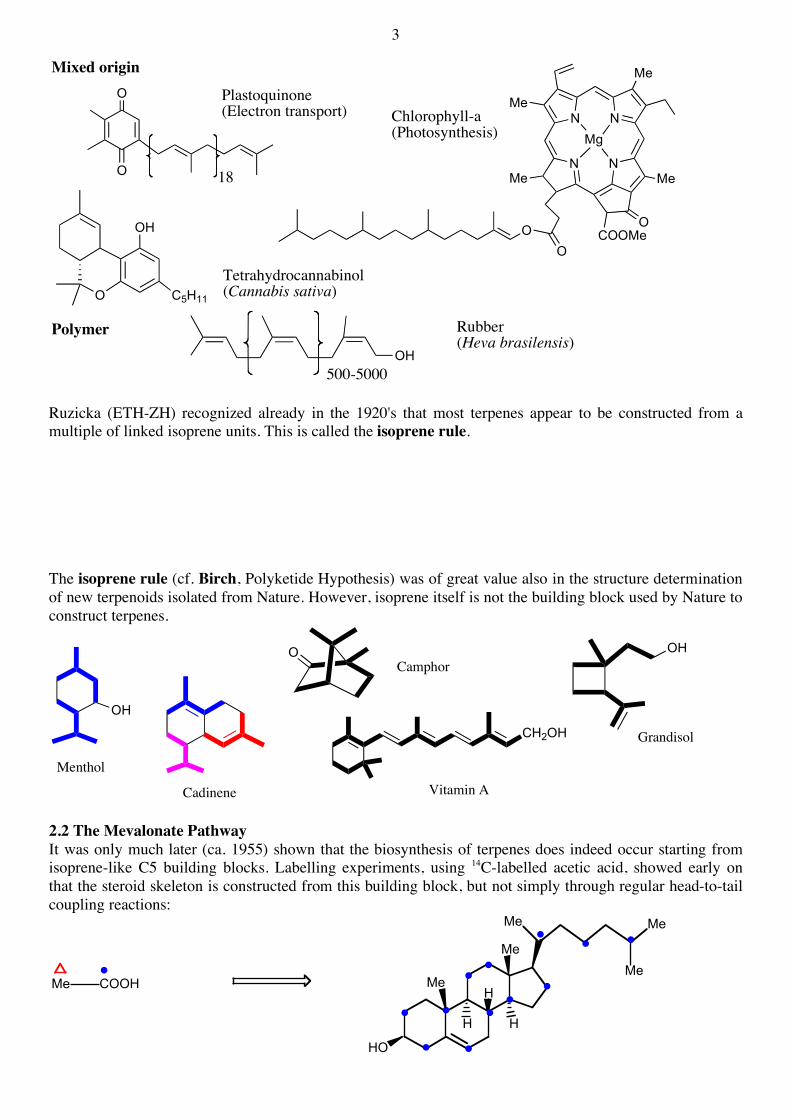
Mixed origin
N N
NN
Me
Me
MeMe
Mg
OCOOMeO
O
Chlorophyll-a(Photosynthesis)
O
O 18
Plastoquinone(Electron transport)
O
OH
C5H11
Tetrahydrocannabinol(Cannabis sativa)
Polymer
OH
Rubber(Heva brasilensis)
500-5000 Ruzicka (ETH-ZH) recognized already in the 1920's that most terpenes appear to be constructed from a multiple of linked isoprene units. This is called the isoprene rule. The isoprene rule (cf. Birch, Polyketide Hypothesis) was of great value also in the structure determination of new terpenoids isolated from Nature. However, isoprene itself is not the building block used by Nature to construct terpenes.
CH2OH
OH
O OH
Vitamin ACadinene
Grandisol
Camphor
Menthol
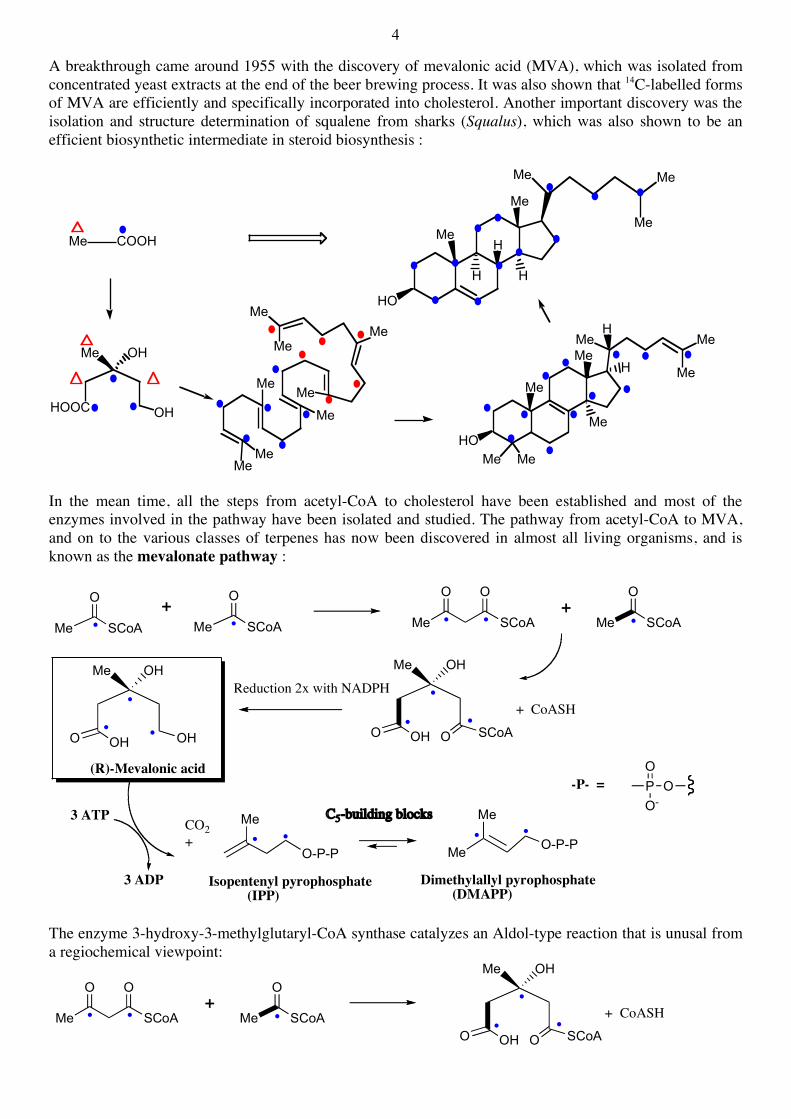
2.2 The Mevalonate Pathway It was only much later (ca. 1955) shown that the biosynthesis of terpenes does indeed occur starting from isoprene-like C5 building blocks. Labelling experiments, using 14C-labelled acetic acid, showed early on that the steroid skeleton is constructed from this building block, but not simply through regular head-to-tail coupling reactions:
Me COOH
HO
MeH
Me
Me Me
Me
HH
4
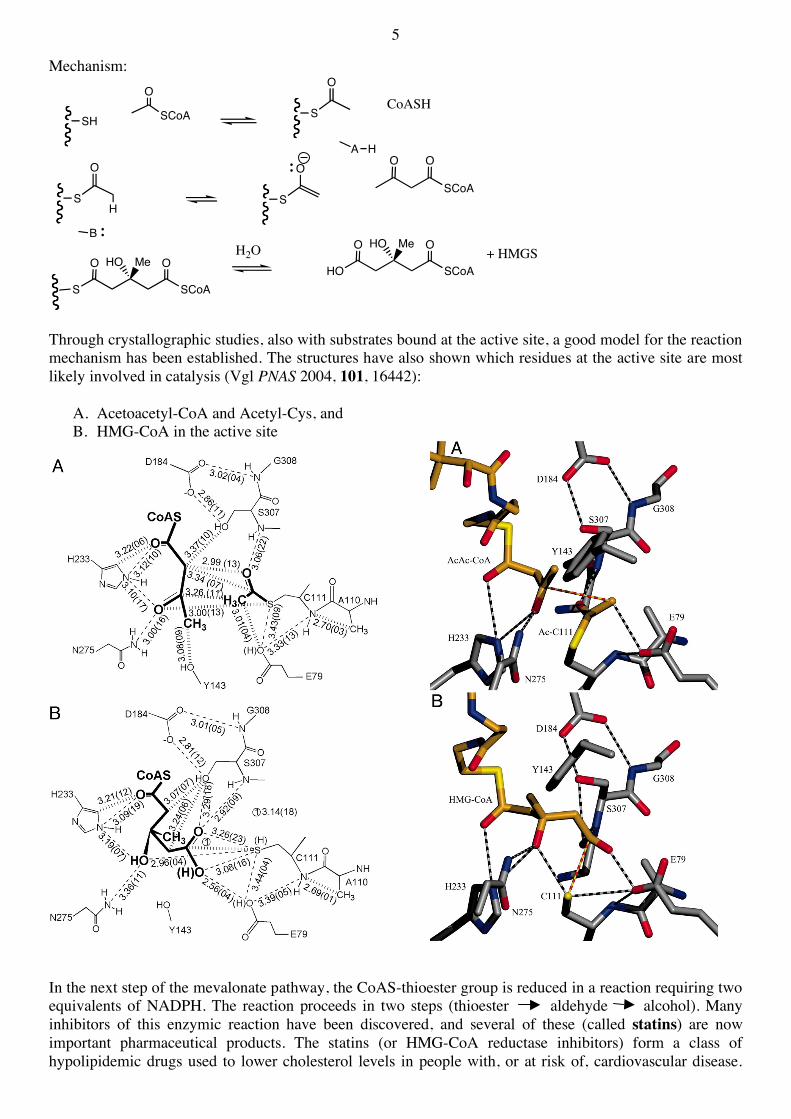
A breakthrough came around 1955 with the discovery of mevalonic acid (MVA), which was isolated from concentrated yeast extracts at the end of the beer brewing process. It was also shown that 14C-labelled forms of MVA are efficiently and specifically incorporated into cholesterol. Another important discovery was the isolation and structure determination of squalene from sharks (Squalus), which was also shown to be an efficient biosynthetic intermediate in steroid biosynthesis :
HO
MeH
Me
Me Me
Me
HH
Me COOH
Me OH
HOOCOH
MeMe
Me
Me
Me
Me
Me
Me Me
Me
Me
Me
HO
Me
MeH
H
Me
Me
In the mean time, all the steps from acetyl-CoA to cholesterol have been established and most of the enzymes involved in the pathway have been isolated and studied. The pathway from acetyl-CoA to MVA, and on to the various classes of terpenes has now been discovered in almost all living organisms, and is known as the mevalonate pathway :
Me
O
SCoA Me
O
SCoA Me SCoA
O O
Me
O
SCoA
Me OH
O OH O SCoA
Me OH
O OH
Me
OH
MeO-P-P
Me
O-P-P
P
O
O-
O
CO2
+
+ CoASH
3 ADP
3 ATP C5-building blocks
Isopentenyl pyrophosphate (IPP)
Dimethylallyl pyrophosphate (DMAPP)
(R)-Mevalonic acid
Reduction 2x with NADPH
++
-P- =
The enzyme 3-hydroxy-3-methylglutaryl-CoA synthase catalyzes an Aldol-type reaction that is unusal from a regiochemical viewpoint:
Me OH
O OH O SCoA
+ CoASHMe SCoA
O O
Me
O
SCoA
+
5
Mechanism:
SHSCoA
O
S
O
CoASH
S
O
H
B
S
O
SCoA
O O
A H
S SCoA
O OHO Me
H2O
HO SCoA
O OHO Me+ HMGS
Through crystallographic studies, also with substrates bound at the active site, a good model for the reaction mechanism has been established. The structures have also shown which residues at the active site are most likely involved in catalysis (Vgl PNAS 2004, 101, 16442):
A. Acetoacetyl-CoA and Acetyl-Cys, and B. HMG-CoA in the active site
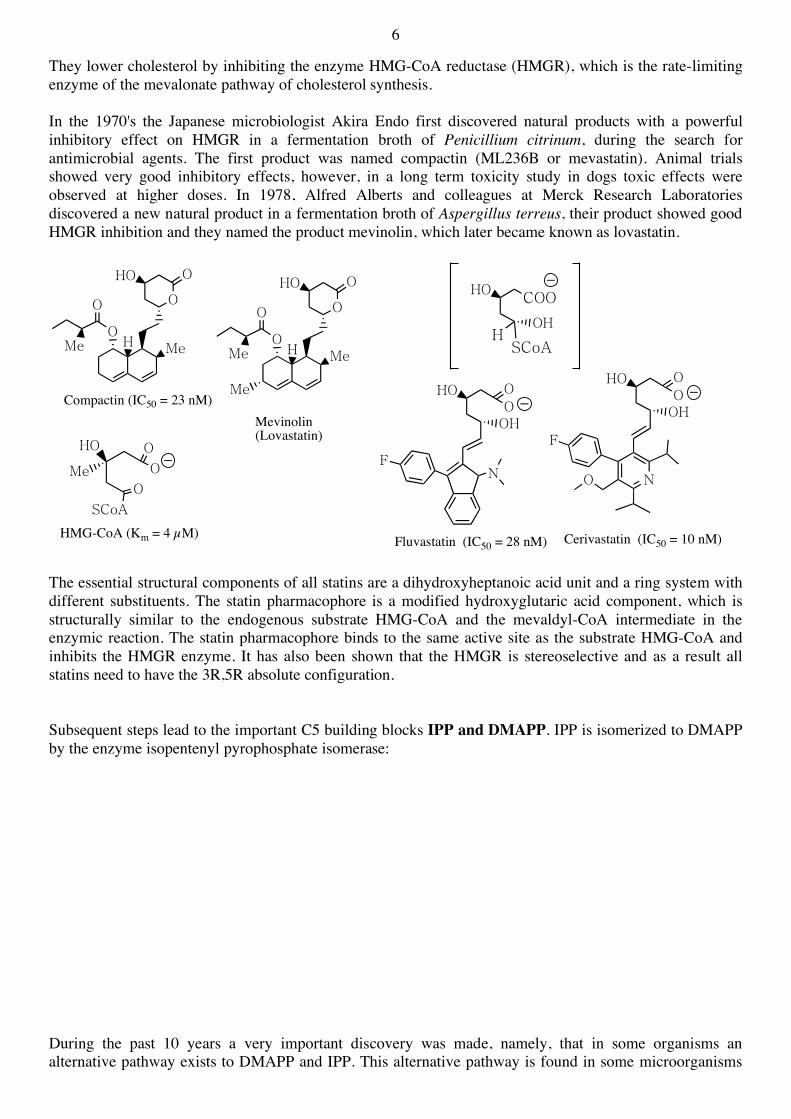
In the next step of the mevalonate pathway, the CoAS-thioester group is reduced in a reaction requiring two equivalents of NADPH. The reaction proceeds in two steps (thioester aldehyde alcohol). Many inhibitors of this enzymic reaction have been discovered, and several of these (called statins) are now important pharmaceutical products. The statins (or HMG-CoA reductase inhibitors) form a class of hypolipidemic drugs used to lower cholesterol levels in people with, or at risk of, cardiovascular disease.
6
They lower cholesterol by inhibiting the enzyme HMG-CoA reductase (HMGR), which is the rate-limiting enzyme of the mevalonate pathway of cholesterol synthesis. In the 1970's the Japanese microbiologist Akira Endo first discovered natural products with a powerful inhibitory effect on HMGR in a fermentation broth of Penicillium citrinum, during the search for antimicrobial agents. The first product was named compactin (ML236B or mevastatin). Animal trials showed very good inhibitory effects, however, in a long term toxicity study in dogs toxic effects were observed at higher doses. In 1978, Alfred Alberts and colleagues at Merck Research Laboratories discovered a new natural product in a fermentation broth of Aspergillus terreus, their product showed good HMGR inhibition and they named the product mevinolin, which later became known as lovastatin.
!
!
"# $
!
$! !
"#
Compactin (IC50 = 23 nM)
$!
"#
!
%&'(
!
!
HMG-CoA (Km = 4 µM)
$! !
!
!$
)
*
Fluvastatin (IC50 = 28 nM)
)!
$! !
!
!$
*
Cerivastatin (IC50 = 10 nM)
!
!
"# $
!
$! !
"#
"#
Mevinolin(Lovastatin)
%&'(
&!!$!
!$$
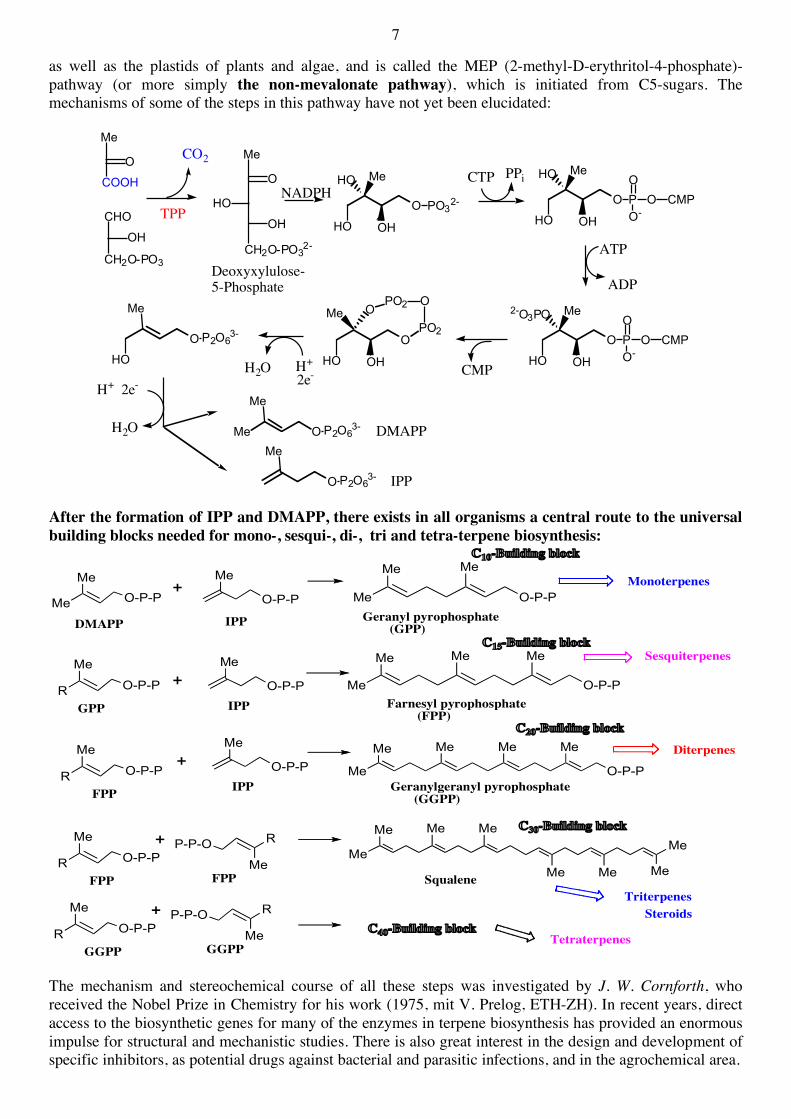
The essential structural components of all statins are a dihydroxyheptanoic acid unit and a ring system with different substituents. The statin pharmacophore is a modified hydroxyglutaric acid component, which is structurally similar to the endogenous substrate HMG-CoA and the mevaldyl-CoA intermediate in the enzymic reaction. The statin pharmacophore binds to the same active site as the substrate HMG-CoA and inhibits the HMGR enzyme. It has also been shown that the HMGR is stereoselective and as a result all statins need to have the 3R,5R absolute configuration. Subsequent steps lead to the important C5 building blocks IPP and DMAPP. IPP is isomerized to DMAPP by the enzyme isopentenyl pyrophosphate isomerase: During the past 10 years a very important discovery was made, namely, that in some organisms an alternative pathway exists to DMAPP and IPP. This alternative pathway is found in some microorganisms
7
as well as the plastids of plants and algae, and is called the MEP (2-methyl-D-erythritol-4-phosphate)-pathway (or more simply the non-mevalonate pathway), which is initiated from C5-sugars. The mechanisms of some of the steps in this pathway have not yet been elucidated:
Me
O
COOH
CHO
OH
CH2O-PO3
Me
O
OH
CH2O-PO32-
HO O PO32-
HO
MeHO
OH
IPP
Deoxyxylulose-5-Phosphate
CO2
TPP
NADPH
O P2O63-
HO
Me
DMAPP
CTP PPi
O P
HO
MeHO
OH
O
O-
O CMP
ATP
ADP
O P
HO
Me2-O3PO
OH
O
O-
O CMP
CMP
O
PO2
HO
Me O
OH
PO2 O
H+2e-
H2O H+
2e-
H2O Me O P2O63-
Me
O P2O63-
Me
After the formation of IPP and DMAPP, there exists in all organisms a central route to the universal building blocks needed for mono-, sesqui-, di-, tri and tetra-terpene biosynthesis:
Me
RO-P-P
Me
O-P-P Me
Me Me
O-P-P
Me
Me
RO-P-P
Me
RP-P-O
Me
Me Me Me
Me
MeMeMe
Me
MeO-P-P
Me
O-P-P Me
Me
R
O-P-P
Me Me
O-P-P
Me
O-P-P Me
Me Me Me
O-P-P
Me
+
C20-Building block
Diterpenes
Geranylgeranyl pyrophosphate (GGPP)
+
Steroids
DMAPP
C30-Building block
C15-Building block
C10-Building block
Triterpenes
Squalene
Sesquiterpenes
Monoterpenes
Farnesyl pyrophosphate (FPP)
+
GPP
Geranyl pyrophosphate (GPP)
+
IPPFPP
IPP
IPP
FPPFPP
TetraterpenesC40-Building block
Me
RO-P-P
Me
RP-P-O
+
GGPPGGPP The mechanism and stereochemical course of all these steps was investigated by J. W. Cornforth, who received the Nobel Prize in Chemistry for his work (1975, mit V. Prelog, ETH-ZH). In recent years, direct access to the biosynthetic genes for many of the enzymes in terpene biosynthesis has provided an enormous impulse for structural and mechanistic studies. There is also great interest in the design and development of specific inhibitors, as potential drugs against bacterial and parasitic infections, and in the agrochemical area.
8
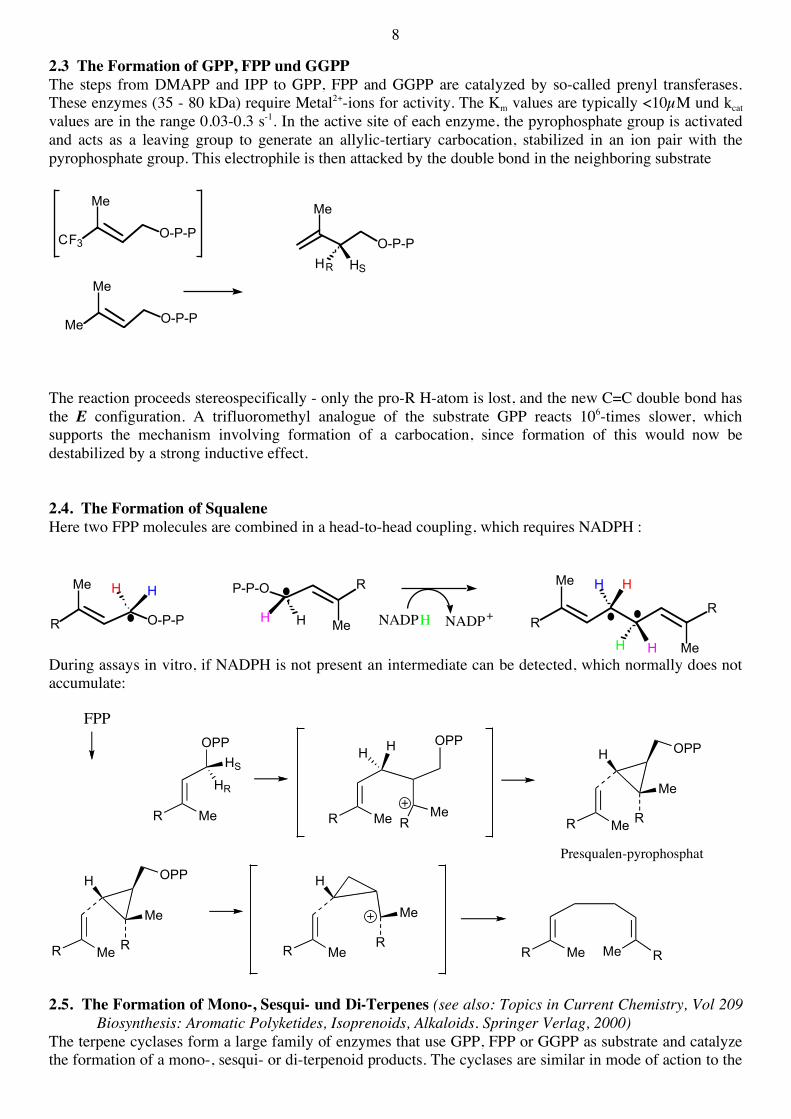
2.3 The Formation of GPP, FPP und GGPP The steps from DMAPP and IPP to GPP, FPP and GGPP are catalyzed by so-called prenyl transferases. These enzymes (35 - 80 kDa) require Metal2+-ions for activity. The Km values are typically <10µM und kcat values are in the range 0.03-0.3 s-1. In the active site of each enzyme, the pyrophosphate group is activated and acts as a leaving group to generate an allylic-tertiary carbocation, stabilized in an ion pair with the pyrophosphate group. This electrophile is then attacked by the double bond in the neighboring substrate
Me
MeO-P-P
Me
O-P-P
HSHR
Me
CF3O-P-P
The reaction proceeds stereospecifically - only the pro-R H-atom is lost, and the new C=C double bond has the E configuration. A trifluoromethyl analogue of the substrate GPP reacts 106-times slower, which supports the mechanism involving formation of a carbocation, since formation of this would now be destabilized by a strong inductive effect. 2.4. The Formation of Squalene Here two FPP molecules are combined in a head-to-head coupling, which requires NADPH :
HH
H HMe
RP-P-O
R
R
Me
Me
Me
R O-P-P
H H
H H
NADPH NADP+
During assays in vitro, if NADPH is not present an intermediate can be detected, which normally does not accumulate:
R Me
OPP
HS
HR
OPP
Me
RMeR
H
OPP
Me
RMeR
H
R Me
HH
RMe
OPP
R Me Me R
Presqualen-pyrophosphat
Me
RMeR
H
FPP
2.5. The Formation of Mono-, Sesqui- und Di-Terpenes (see also: Topics in Current Chemistry, Vol 209 Biosynthesis: Aromatic Polyketides, Isoprenoids, Alkaloids. Springer Verlag, 2000) The terpene cyclases form a large family of enzymes that use GPP, FPP or GGPP as substrate and catalyze the formation of a mono-, sesqui- or di-terpenoid products. The cyclases are similar in mode of action to the
9
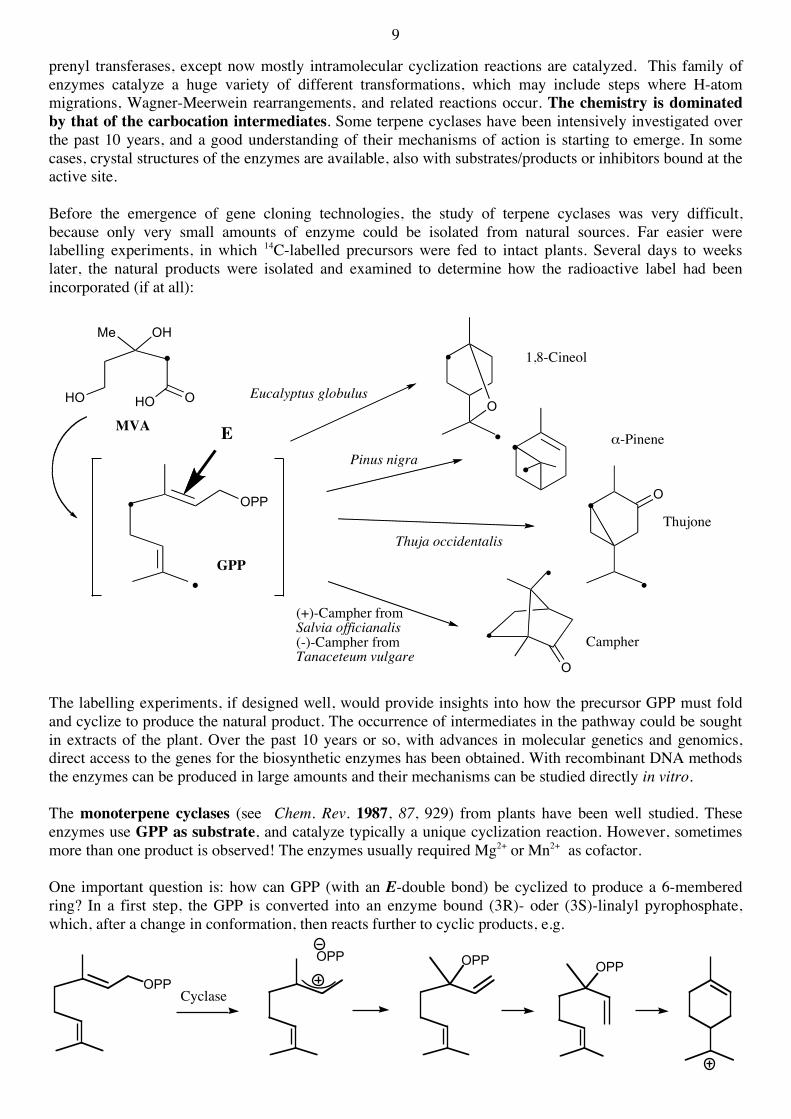
prenyl transferases, except now mostly intramolecular cyclization reactions are catalyzed. This family of enzymes catalyze a huge variety of different transformations, which may include steps where H-atom migrations, Wagner-Meerwein rearrangements, and related reactions occur. The chemistry is dominated by that of the carbocation intermediates. Some terpene cyclases have been intensively investigated over the past 10 years, and a good understanding of their mechanisms of action is starting to emerge. In some cases, crystal structures of the enzymes are available, also with substrates/products or inhibitors bound at the active site. Before the emergence of gene cloning technologies, the study of terpene cyclases was very difficult, because only very small amounts of enzyme could be isolated from natural sources. Far easier were labelling experiments, in which 14C-labelled precursors were fed to intact plants. Several days to weeks later, the natural products were isolated and examined to determine how the radioactive label had been incorporated (if at all):
Me OH
HO OHO O
OOPP
O
Thuja occidentalis
Thujone
!-Pinene
1,8-Cineol
Pinus nigra
Eucalyptus globulus
Campher
(+)-Campher fromSalvia officianalis(-)-Campher fromTanaceteum vulgare
GPP
EMVA
The labelling experiments, if designed well, would provide insights into how the precursor GPP must fold and cyclize to produce the natural product. The occurrence of intermediates in the pathway could be sought in extracts of the plant. Over the past 10 years or so, with advances in molecular genetics and genomics, direct access to the genes for the biosynthetic enzymes has been obtained. With recombinant DNA methods the enzymes can be produced in large amounts and their mechanisms can be studied directly in vitro. The monoterpene cyclases (see Chem. Rev. 1987, 87, 929) from plants have been well studied. These enzymes use GPP as substrate, and catalyze typically a unique cyclization reaction. However, sometimes more than one product is observed! The enzymes usually required Mg2+ or Mn2+ as cofactor. One important question is: how can GPP (with an E-double bond) be cyclized to produce a 6-membered ring? In a first step, the GPP is converted into an enzyme bound (3R)- oder (3S)-linalyl pyrophosphate, which, after a change in conformation, then reacts further to cyclic products, e.g.
OPP
OPP OPPOPP
Cyclase
10
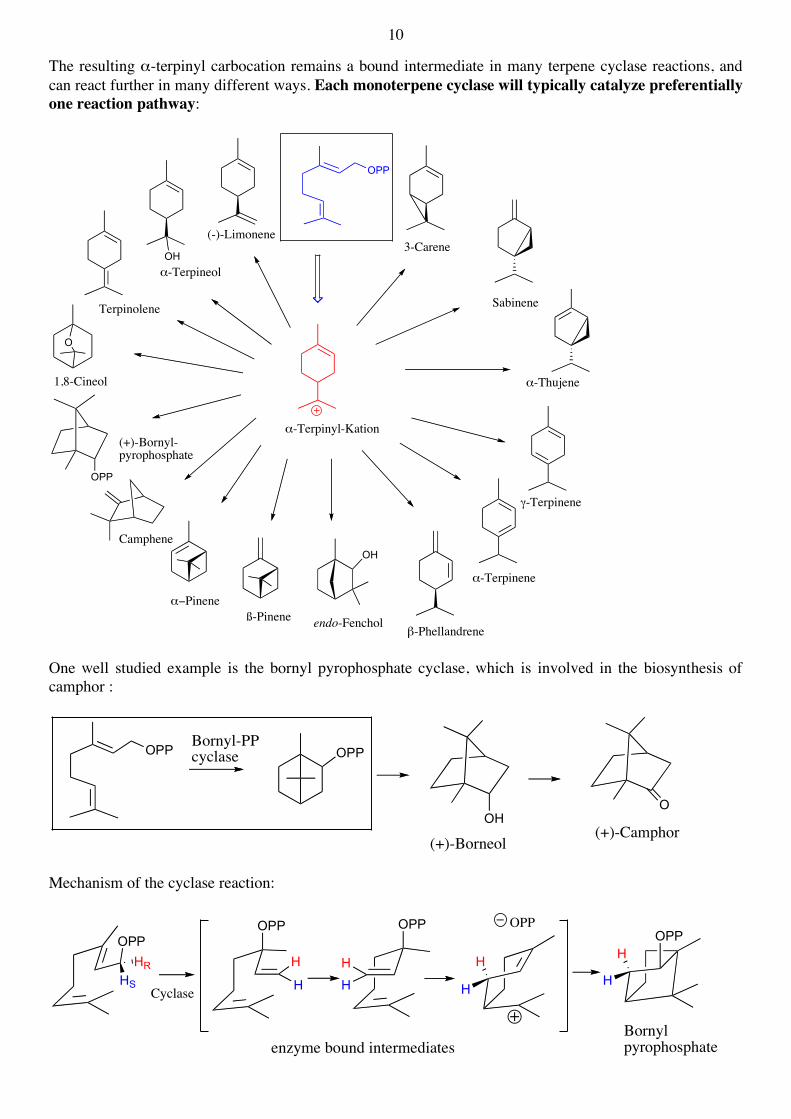
The resulting α-terpinyl carbocation remains a bound intermediate in many terpene cyclase reactions, and can react further in many different ways. Each monoterpene cyclase will typically catalyze preferentially one reaction pathway:
OPP
3-Carene
Sabinene
!-Thujene
"-Terpinene
!-Terpinene
#-Phellandrene
OH
endo-Fencholß-Pinene
!$Pinene
Camphene
OPP
(+)-Bornyl-pyrophosphate
O
1,8-Cineol
OH
!-Terpineol
(-)-Limonene
Terpinolene
!-Terpinyl-Kation
One well studied example is the bornyl pyrophosphate cyclase, which is involved in the biosynthesis of camphor :
OPP OPP
O
OH
(+)-Camphor(+)-Borneol
Bornyl-PP cyclase
Mechanism of the cyclase reaction:
OPP
OPP OPPOPP
Cyclase
!
enzyme bound intermediates
HS
HR H
H
H
H
H
H
Bornylpyrophosphate
H
H
OPP
11
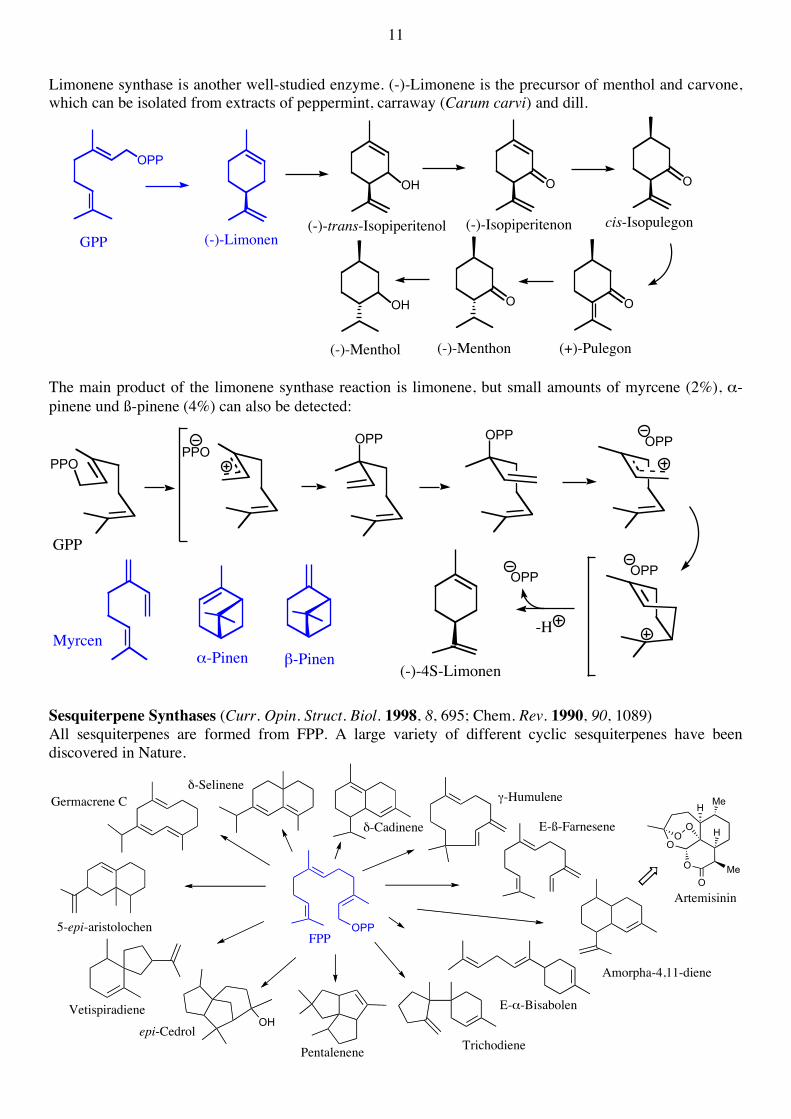
Limonene synthase is another well-studied enzyme. (-)-Limonene is the precursor of menthol and carvone, which can be isolated from extracts of peppermint, carraway (Carum carvi) and dill.
OPP
OH O O
OOOH
GPP (-)-Limonen(-)-trans-Isopiperitenol (-)-Isopiperitenon cis-Isopulegon
(+)-Pulegon(-)-Menthon(-)-Menthol The main product of the limonene synthase reaction is limonene, but small amounts of myrcene (2%), α-pinene und ß-pinene (4%) can also be detected:
PPO
GPP
PPO
OPP OPPOPP
OPP
(-)-4S-Limonen
OPP
-HMyrcen
!-Pinen "-Pinen
Sesquiterpene Synthases (Curr. Opin. Struct. Biol. 1998, 8, 695; Chem. Rev. 1990, 90, 1089) All sesquiterpenes are formed from FPP. A large variety of different cyclic sesquiterpenes have been discovered in Nature.
OPP
!-Cadinene
"-Humulene
E-ß-Farnesene
E-#-Bisabolen
TrichodienePentalenene
OHepi-Cedrol
Vetispiradiene
5-epi-aristolochen
Germacrene C
!-Selinene
FPP
Amorpha-4,11-diene
O
HMe
H
O
OMe
O
O
Artemisinin
12
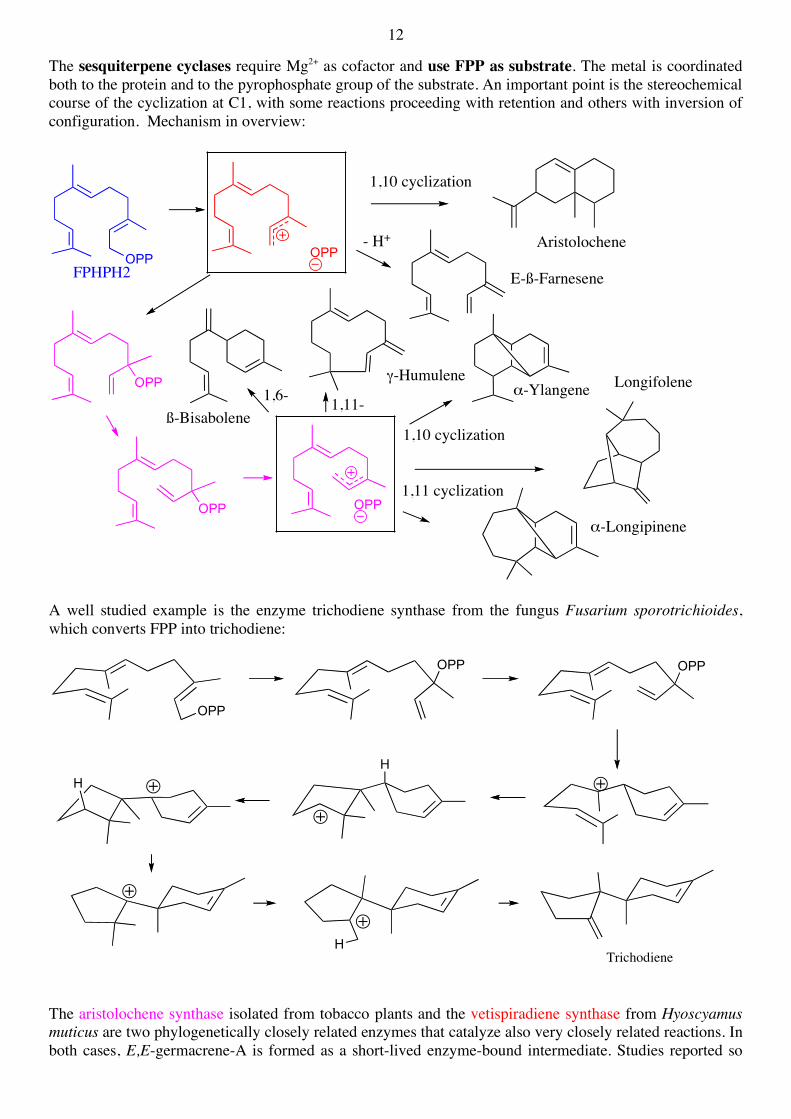
The sesquiterpene cyclases require Mg2+ as cofactor and use FPP as substrate. The metal is coordinated both to the protein and to the pyrophosphate group of the substrate. An important point is the stereochemical course of the cyclization at C1, with some reactions proceeding with retention and others with inversion of configuration. Mechanism in overview:
OPPOPP
OPP
OPP OPP
FPHPH2 E-ß-Farnesene
1,10 cyclization
Aristolochene- H+
!-Humulene Longifolene
"-Longipinene
"-Ylangene
1,11 cyclization
1,10 cyclizationß-Bisabolene
1,6-1,11-
A well studied example is the enzyme trichodiene synthase from the fungus Fusarium sporotrichioides, which converts FPP into trichodiene:
OPP
OPP OPP
H
!
!
Trichodiene
!H
!
H
!
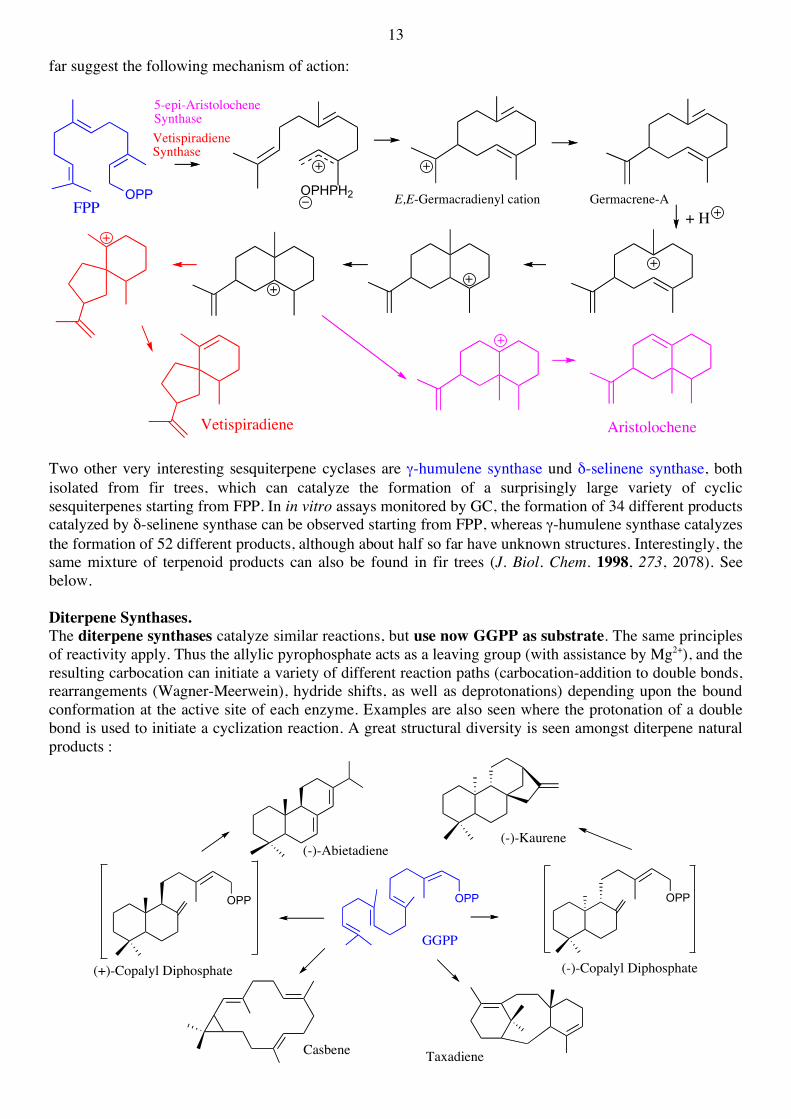
The aristolochene synthase isolated from tobacco plants and the vetispiradiene synthase from Hyoscyamus muticus are two phylogenetically closely related enzymes that catalyze also very closely related reactions. In both cases, E,E-germacrene-A is formed as a short-lived enzyme-bound intermediate. Studies reported so
13
far suggest the following mechanism of action:
OPP E,E-Germacradienyl cationOPHPH2
+ H
AristolocheneVetispiradiene
Germacrene-A
VetispiradieneSynthase
5-epi-AristolocheneSynthase
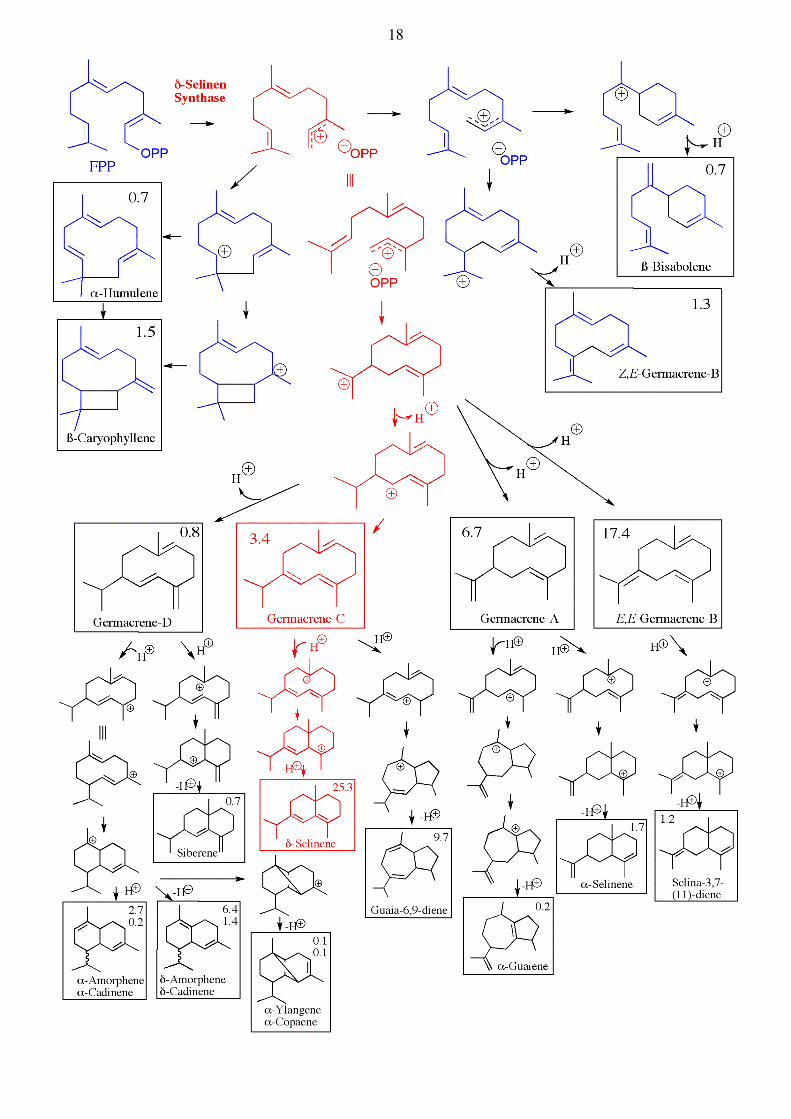
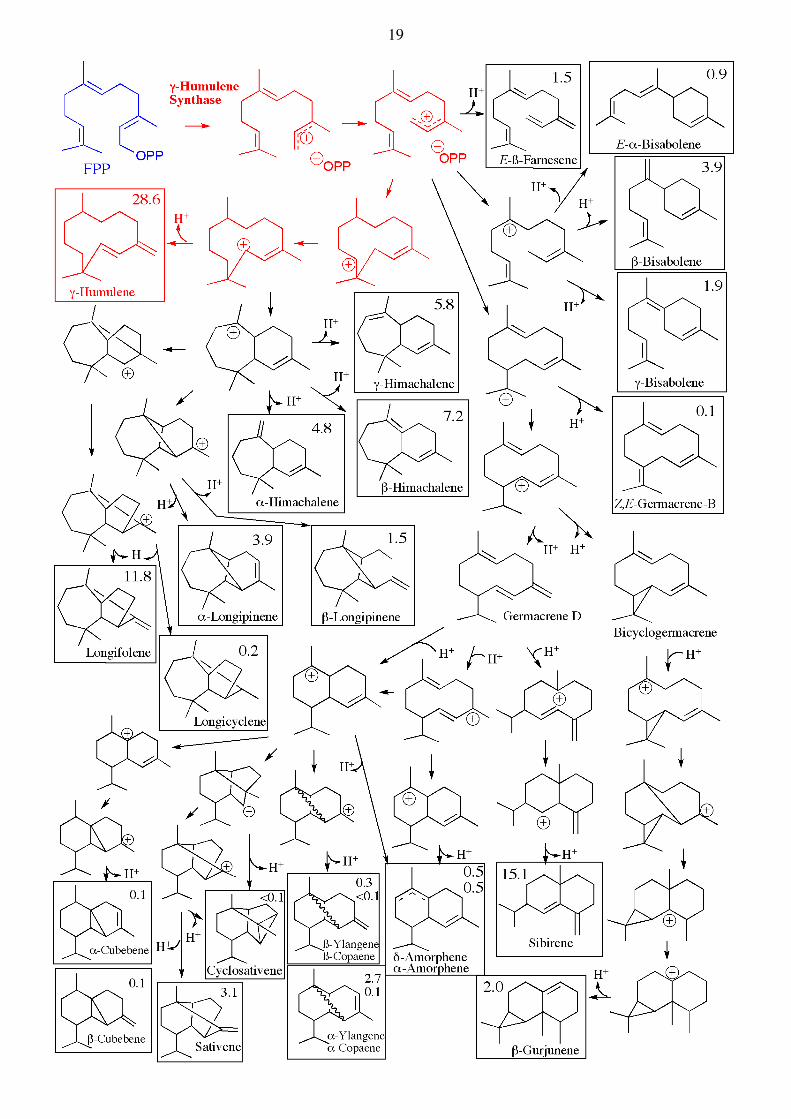
FPP
Two other very interesting sesquiterpene cyclases are γ-humulene synthase und δ-selinene synthase, both isolated from fir trees, which can catalyze the formation of a surprisingly large variety of cyclic sesquiterpenes starting from FPP. In in vitro assays monitored by GC, the formation of 34 different products catalyzed by δ-selinene synthase can be observed starting from FPP, whereas γ-humulene synthase catalyzes the formation of 52 different products, although about half so far have unknown structures. Interestingly, the same mixture of terpenoid products can also be found in fir trees (J. Biol. Chem. 1998, 273, 2078). See below. Diterpene Synthases. The diterpene synthases catalyze similar reactions, but use now GGPP as substrate. The same principles of reactivity apply. Thus the allylic pyrophosphate acts as a leaving group (with assistance by Mg2+), and the resulting carbocation can initiate a variety of different reaction paths (carbocation-addition to double bonds, rearrangements (Wagner-Meerwein), hydride shifts, as well as deprotonations) depending upon the bound conformation at the active site of each enzyme. Examples are also seen where the protonation of a double bond is used to initiate a cyclization reaction. A great structural diversity is seen amongst diterpene natural products :
OPP
GGPP
OPP OPP
(-)-Abietadiene
(+)-Copalyl Diphosphate (-)-Copalyl Diphosphate
(-)-Kaurene
TaxadieneCasbene
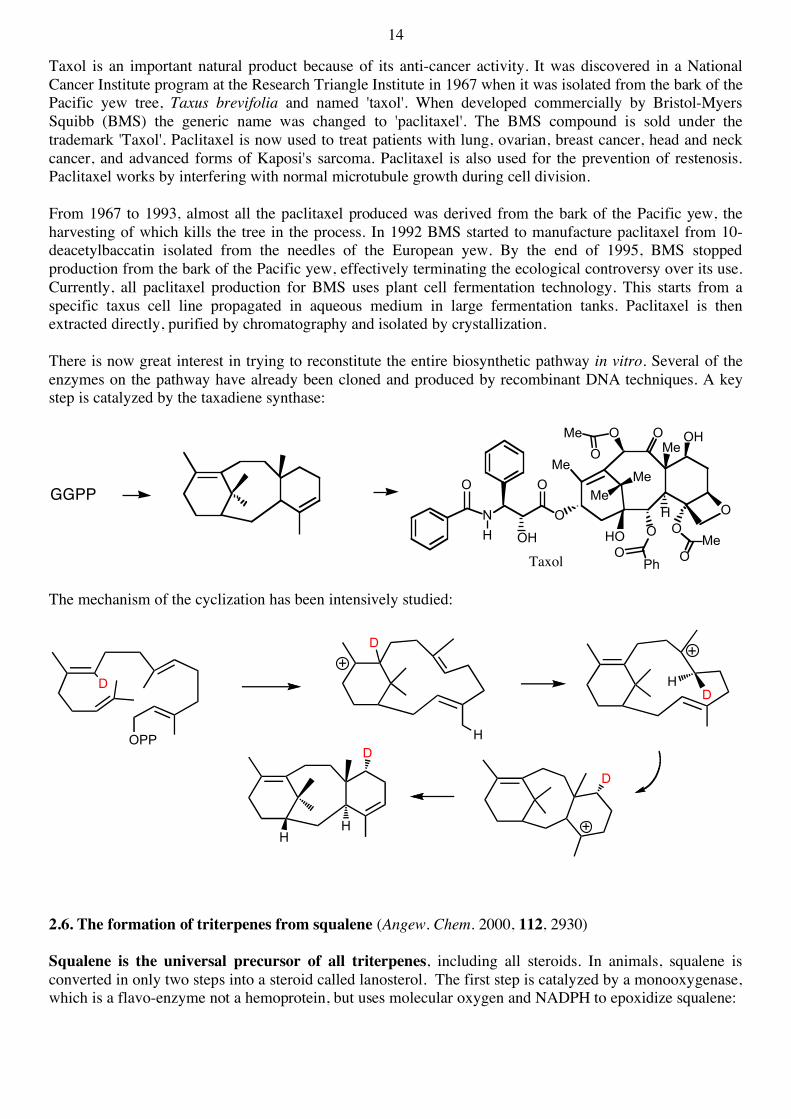
14
Taxol is an important natural product because of its anti-cancer activity. It was discovered in a National Cancer Institute program at the Research Triangle Institute in 1967 when it was isolated from the bark of the Pacific yew tree, Taxus brevifolia and named 'taxol'. When developed commercially by Bristol-Myers Squibb (BMS) the generic name was changed to 'paclitaxel'. The BMS compound is sold under the trademark 'Taxol'. Paclitaxel is now used to treat patients with lung, ovarian, breast cancer, head and neck cancer, and advanced forms of Kaposi's sarcoma. Paclitaxel is also used for the prevention of restenosis. Paclitaxel works by interfering with normal microtubule growth during cell division. From 1967 to 1993, almost all the paclitaxel produced was derived from the bark of the Pacific yew, the harvesting of which kills the tree in the process. In 1992 BMS started to manufacture paclitaxel from 10-deacetylbaccatin isolated from the needles of the European yew. By the end of 1995, BMS stopped production from the bark of the Pacific yew, effectively terminating the ecological controversy over its use. Currently, all paclitaxel production for BMS uses plant cell fermentation technology. This starts from a specific taxus cell line propagated in aqueous medium in large fermentation tanks. Paclitaxel is then extracted directly, purified by chromatography and isolated by crystallization. There is now great interest in trying to reconstitute the entire biosynthetic pathway in vitro. Several of the enzymes on the pathway have already been cloned and produced by recombinant DNA techniques. A key step is catalyzed by the taxadiene synthase:
O
N
H OH
O
O
Me
Me
Me
O O
MeOH
O
O
H
HOO
O
Me
O
MeO
PhTaxol
GGPP
The mechanism of the cyclization has been intensively studied:
OPPH
D
DD
H
D
D
HH
2.6. The formation of triterpenes from squalene (Angew. Chem. 2000, 112, 2930) Squalene is the universal precursor of all triterpenes, including all steroids. In animals, squalene is converted in only two steps into a steroid called lanosterol. The first step is catalyzed by a monooxygenase, which is a flavo-enzyme not a hemoprotein, but uses molecular oxygen and NADPH to epoxidize squalene:
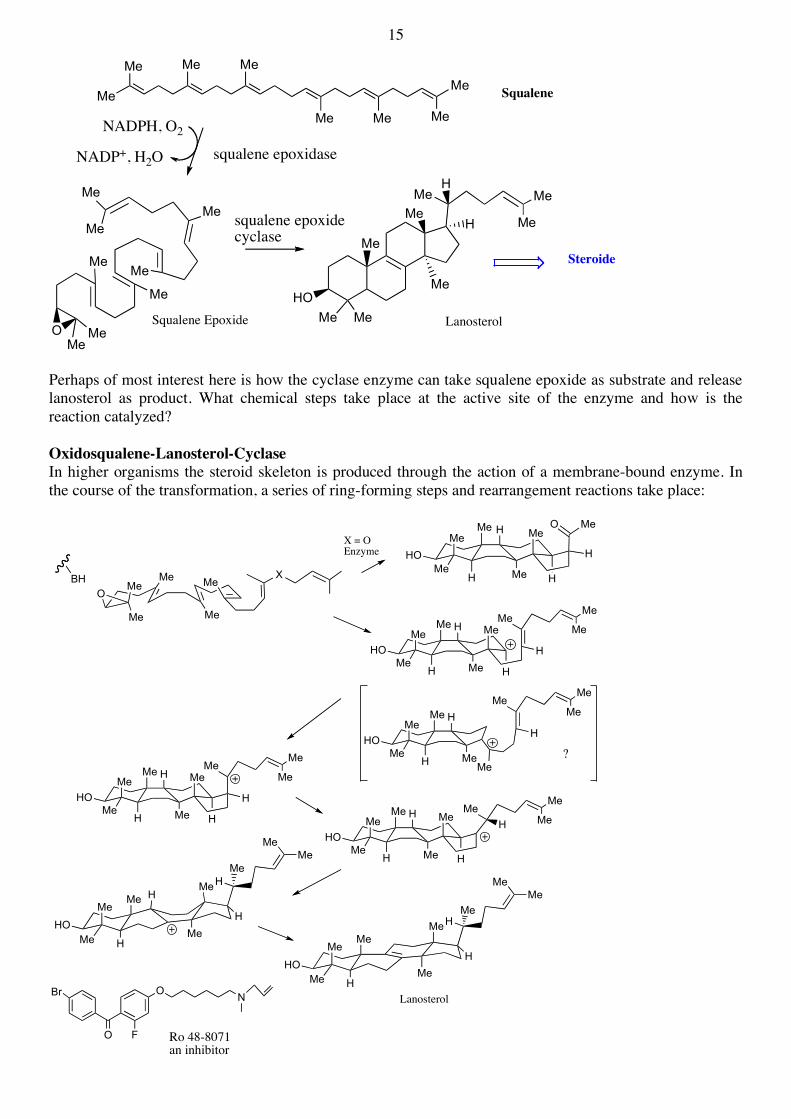
15
O MeMe
Me
Me
Me
Me
Me
Me
Me Me
Me
Me
Me
Me
HO
Me
MeH
H
Squalene Epoxide Lanosterol
Steroide
Me
Me Me Me
Me
MeMeMe
Squalene
NADPH, O2
NADP+, H2O squalene epoxidase
squalene epoxidecyclase
Perhaps of most interest here is how the cyclase enzyme can take squalene epoxide as substrate and release lanosterol as product. What chemical steps take place at the active site of the enzyme and how is the reaction catalyzed? Oxidosqualene-Lanosterol-Cyclase In higher organisms the steroid skeleton is produced through the action of a membrane-bound enzyme. In the course of the transformation, a series of ring-forming steps and rearrangement reactions take place:
Me
Me
O
Me
Me
Me
Me
MeMe
MeMe
Me
HO
Me H
MeH H
MeMe
Me
HO
Me
Me
H
Me
H
Me
Me
H
Me
H
MeO
Me
Me
HO
Me H
MeH H
Lanosterol
X = OEnzyme
BHX
H
Br
O F
ON
Ro 48-8071an inhibitor
MeMe
Me
Me
Me
HO
Me H
MeH
H
Me
Me
MeMe
MeMe
Me
HO
Me H
MeH H
H
MeMe
Me
MeMe
Me
HO
Me H
MeH H
H
MeMe
Me
HO
Me
Me
H
Me
H
Me
Me
H
H
?
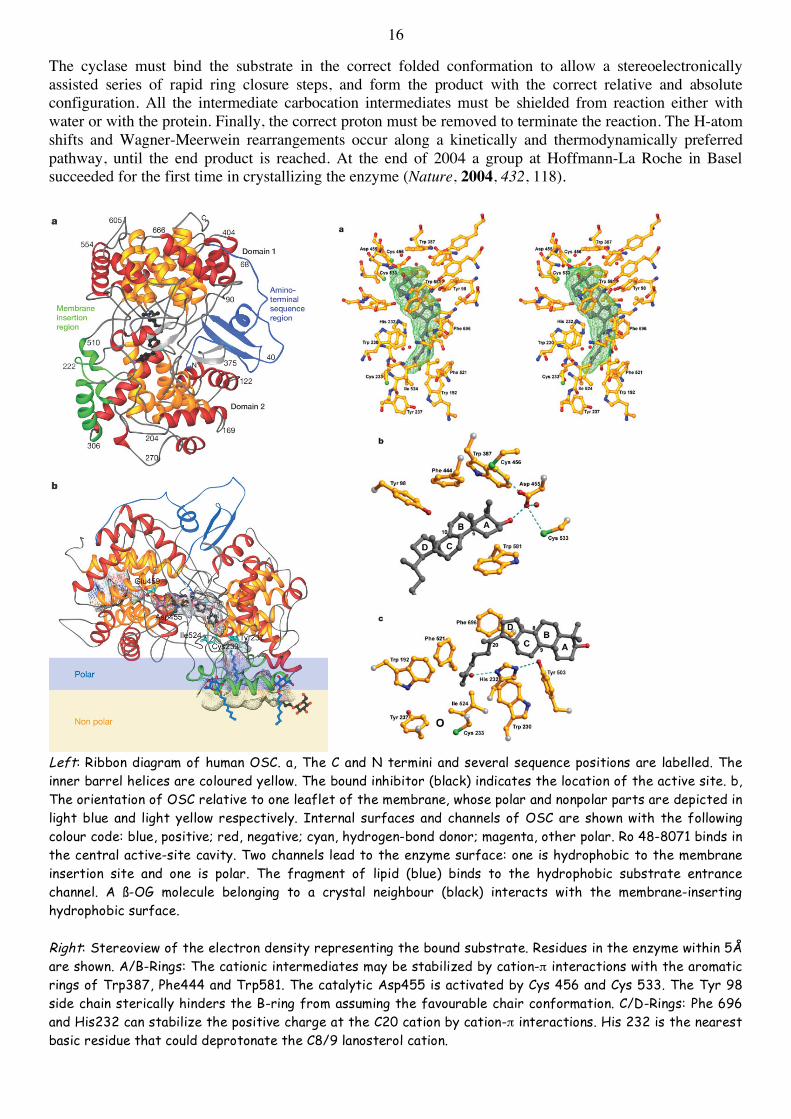
16
The cyclase must bind the substrate in the correct folded conformation to allow a stereoelectronically assisted series of rapid ring closure steps, and form the product with the correct relative and absolute configuration. All the intermediate carbocation intermediates must be shielded from reaction either with water or with the protein. Finally, the correct proton must be removed to terminate the reaction. The H-atom shifts and Wagner-Meerwein rearrangements occur along a kinetically and thermodynamically preferred pathway, until the end product is reached. At the end of 2004 a group at Hoffmann-La Roche in Basel succeeded for the first time in crystallizing the enzyme (Nature, 2004, 432, 118).
Left: Ribbon diagram of human OSC. a, The C and N termini and several sequence positions are labelled. The inner barrel helices are coloured yellow. The bound inhibitor (black) indicates the location of the active site. b, The orientation of OSC relative to one leaflet of the membrane, whose polar and nonpolar parts are depicted in light blue and light yellow respectively. Internal surfaces and channels of OSC are shown with the following colour code: blue, positive; red, negative; cyan, hydrogen-bond donor; magenta, other polar. Ro 48-8071 binds in the central active-site cavity. Two channels lead to the enzyme surface: one is hydrophobic to the membrane insertion site and one is polar. The fragment of lipid (blue) binds to the hydrophobic substrate entrance channel. A ß-OG molecule belonging to a crystal neighbour (black) interacts with the membrane-inserting hydrophobic surface. Right: Stereoview of the electron density representing the bound substrate. Residues in the enzyme within 5Å are shown. A/B-Rings: The cationic intermediates may be stabilized by cation-π interactions with the aromatic rings of Trp387, Phe444 and Trp581. The catalytic Asp455 is activated by Cys 456 and Cys 533. The Tyr 98 side chain sterically hinders the B-ring from assuming the favourable chair conformation. C/D-Rings: Phe 696 and His232 can stabilize the positive charge at the C20 cation by cation-π interactions. His 232 is the nearest basic residue that could deprotonate the C8/9 lanosterol cation.
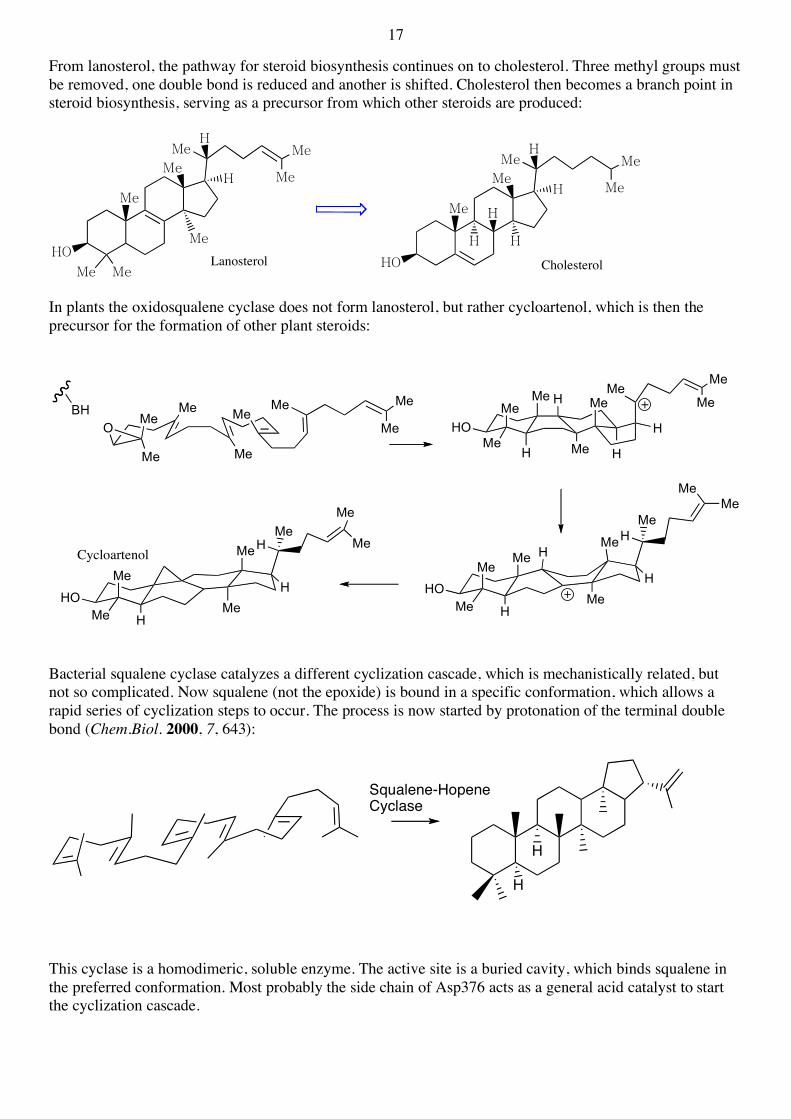
17
From lanosterol, the pathway for steroid biosynthesis continues on to cholesterol. Three methyl groups must be removed, one double bond is reduced and another is shifted. Cholesterol then becomes a branch point in steroid biosynthesis, serving as a precursor from which other steroids are produced:
!"
!" !"
!"
!"
!"
#$
!"
!"#
#
Lanosterol
!" !"
!"
#$
!"
!"#
#
Cholesterol
##
#
In plants the oxidosqualene cyclase does not form lanosterol, but rather cycloartenol, which is then the precursor for the formation of other plant steroids:
Me
Me
O
Me
Me
MeBHMe Me
Me
Me
MeMe
MeMe
Me
HO
Me H
MeH H
H
MeMe
Me
HO
Me
Me
H
Me
H
Me
Me
H
H
Me
Me
HO
Me
Me
H
Me
H
Me
Me
H
Cycloartenol
Bacterial squalene cyclase catalyzes a different cyclization cascade, which is mechanistically related, but not so complicated. Now squalene (not the epoxide) is bound in a specific conformation, which allows a rapid series of cyclization steps to occur. The process is now started by protonation of the terminal double bond (Chem.Biol. 2000, 7, 643):
H
H
Squalene-HopeneCyclase
This cyclase is a homodimeric, soluble enzyme. The active site is a buried cavity, which binds squalene in the preferred conformation. Most probably the side chain of Asp376 acts as a general acid catalyst to start the cyclization cascade.
18
19